
RESUMEN
Tras la actualización de la regulación europea sobre dispositivos médicos de 2017 y su posterior entrada en vigor en 2021, persisten multitud de dudas en cuanto a su implementación práctica en la investigación en productos sanitarios (PS) en España. Desarrollar PS implica la colaboración de multitud de actores, incluyendo investigadores, comités de ética, organismos notificados, Agencia Española de Medicamentos y Productos Sanitarios, industria de dispositivos y diversos colaboradores, que se han visto obligados a adaptarse a esta nueva regulación. Este hecho ha requerido un esfuerzo y un consumo de recursos significativo para comprender y aplicar las distintas directrices que persiguen unificar los protocolos en PS y asegurar la seguridad y la eficacia de estos productos. Desde su implementación, la colaboración entre las distintas partes ha permitido progresar y entender las nuevas dificultades que representa esta normativa. El objetivo de este documento es definir de manera práctica el contenido de dicha regulación y exponer las perspectivas de los diferentes actores implicados en el desarrollo de PS.
Palabras clave: Producto sanitario. Investigación sanitaria. Regulación en investigación.
ABSTRACT
Despite the 2017 update to European medical device regulation and its entry into force in 2021, many doubts persist about its real-world application in medical device (MD) research in Spain. The development of MDs requires collaboration among numerous stakeholders, including researchers, ethics committees, notified bodies, the Spanish Agency of Medicines and Medical Devices, manufacturers, and other involved parties—all of whom have had to adapt to the new regulatory framework. This has required a significant effort and consumption of resources to understand and apply the different clinical practice guidelines to unify protocols in MD research and ensure the safety and efficacy profile of these products. Although challenges remain, the adoption of this legislation has fostered collaboration among stakeholders, enabling progress and a better understanding of the new obstacles it presents. The aim of this review is to define, in a practical way, the content of this regulation and expose the perspectives of the different actors involved in the development of MD.
Keywords: Medical device. Health research. Research regulation.
Abreviaturas
AEMPS: Agencia Española de Medicamentos y Productos Sanitarios. CE: Certificación Europea. CEIm: comité de ética de la investigación con medicamentos. EU-MDR: Reglamento Europeo de Productos Sanitarios. MDR: Reglamento de Productos Sanitarios. ON: organismo notificado. PS: producto sanitario.
INTRODUCCIÓN
Desde la actualización del Reglamento Europeo de Productos Sanitarios (PS) en 2017 con el Reglamento (EU) 2017/745 de Productos Sanitarios (EU-MDR)1 y el reglamento (EU) 2017/746 de productos in vitro (EU-IVDR)2, así como con la publicación en España del Real Decreto 192/2023 de 21 de marzo3, ha surgido la necesidad de adaptarse a las nuevas normativas impulsadas desde Europa. El objetivo que persiguen estas regulaciones es establecer un nuevo marco normativo sólido, transparente, previsible y sostenible para los PS, que garantice los más altos niveles de seguridad y de protección de la salud de pacientes y usuarios, y que asimismo impulse la innovación y los intereses de las pequeñas y medianas empresas (PYME) que desarrollan sus actividades en este sector, obteniendo de esta manera el marcado de Certificación Europea (CE). Este reglamento armoniza las normas aplicables a la introducción en el mercado y la puesta en servicio en la Unión Europea de PS y sus accesorios, permitiendo así que estos se acojan al principio de libre circulación de mercancías y garantizando, además, un nivel de protección elevado, de modo que los productos en circulación no presenten riesgos para la salud de los pacientes, usuarios o terceras personas, y alcancen las prestaciones asignadas por el fabricante, cuando se utilicen en las condiciones previstas3.
La complejidad de esta nueva normativa ha llevado a que desde el punto de vista académico se hayan publicado documentos previos, en los ámbitos tanto europeo como nacionales, para ayudar a comprender las nuevas regulaciones4,5. No obstante, estos cambios regulatorios generan, de manera invariable, tensiones entre los distintos actores implicados en el desarrollo de PS. Entre ellos se encuentran los organismos reguladores, como son la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS), los comités éticos de la investigación con medicamentos (CEIm) y los organismos notificados (ON); la industria, compuesta por distintas grandes empresas, PYME y empresas emergentes (start-up); los colaboradores, como pueden ser empresas encargadas del registro y el análisis de datos sanitarios, o distintos tipos de asesorías; los investigadores y, finalmente, los propios pacientes y sus asociaciones. Dado que todos estos actores persiguen un mismo objetivo común, la colaboración entre cada una de las partes en la comprensión de esta regulación, aportando su perspectiva única y las dificultades que individualmente presentan, puede llevar a mejorar el desarrollo de PS y facilitar un proceso que cada vez es más complejo (figura 1).

Figura 1. Figura central. Partes interesadas en el desarrollo de productos sanitarios, y retos y amenazas a los que se enfrentan. Los iconos han sido realizados por Freepik, Smalllikeart, Rizal2109, e Iconpro en www.flaticon.com. Imagen obtenida de Servier Medical Art, bajo licencia CC BY 4.0 (https://smart.servier.com).
El objetivo de este documento es resumir el contenido de la EU-MDR para facilitar su comprensión y exponer la perspectiva de cada una de las partes implicadas en el proceso de desarrollo de PS. En enero de 2025, con el objetivo mencionado, se organizó una reunión presencial impulsada por la Fundación Epic en la que se expusieron estas perspectivas con distintas presentaciones. A continuación, se resume el contenido de las presentaciones, las cuestiones y los debates que tuvieron lugar en la reunión.
Definiciones y nomenclatura en productos sanitarios
A pesar de que en el ámbito médico la utilización de PS es ubicua, el personal sanitario raras veces está familiarizado con la nomenclatura y las definiciones que se aplican a estos. En la tabla 1 se resumen de manera breve los principales términos que se utilizarán en este documento. El EU-MDR define claramente la terminología a utilizar en su artículo 21, pero resulta conveniente extenderse en algunas definiciones para comprender totalmente los matices del desarrollo de PS. La clasificación de riesgo de los PS tiene en consideración el propósito de uso del dispositivo y sus riesgos inherentes (EU-MDR, anexo VIII1, y guía de la Comisión Europea6). Los dispositivos pueden clasificarse como de bajo riesgo (clase I), medio riesgo (clase IIa) y alto riesgo (clase IIb y clase III), y en el caso de los productos sanitarios para diagnóstico in vitro en las clases A, B, C y D, siendo la clase A la de menor riesgo y la clase D la de mayor riesgo. Los dispositivos de clase I que se consideran más simples pueden ser autocertificados por el fabricante, pero otros dispositivos de clase I (que sean estériles o que tengan función de medición) y los de las clases IIa, IIb y III deben superar una valoración de conformidad que implica la participación de un ON. Los dispositivos implantables y los que estén en contacto directo con el sistema circulatorio se definen todos como de alto riesgo (clase III); de estos, el 40% son de aplicación cardiovascular7. En la figura 2 se presentan algunos ejemplos de las distintas clases de dispositivos. Se definen como:
Tabla 1. Definiciones de los principales términos utilizados en el documento
| Término | Definición |
|---|---|
| Producto sanitario | Todo instrumento, dispositivo, equipo, programa informático, implante, reactivo, material u otro artículo destinado por el fabricante a ser utilizado en personas, por separado o en combinación, con alguna de las siguientes finalidades médicas específicas:
– Diagnóstico, prevención, seguimiento, predicción, pronóstico, tratamiento o alivio de una enfermedad. – Diagnóstico, seguimiento, tratamiento, alivio o compensación de una lesión o de una discapacidad. – Investigación, sustitución o modificación de la anatomía de un proceso o estado fisiológico o patológico. – Obtención de información mediante el examen in vitro de muestras procedentes del cuerpo humano, incluyendo donaciones de órganos, sangre y tejidos. |
| Además, no ejerce su acción principal prevista en el interior o en la superficie del cuerpo humano por mecanismos farmacológicos, inmunológicos ni metabólicos, pero a su función puedan contribuir tales mecanismos.
Los siguientes también se considerarán productos sanitarios: – Productos de control o apoyo de la concepción. – Productos destinados específicamente a la limpieza, desinfección o esterilización de los productos que se contemplan en el artículo 1, apartado 4, y en el párrafo primero del presente punto. (MDR, artículo 2.1) |
|
| Producto de diagnóstico in vitro | Cualquier producto sanitario que consista en un reactivo, producto reactivo, calibrador, material de control, kit, instrumento, aparato, pieza de equipo, programa informático o sistema, utilizado solo o en combinación, destinado por el fabricante a ser utilizado in vitro para el estudio de muestras procedentes del cuerpo humano, incluidas las donaciones de sangre y tejidos, única o principalmente con el fin de proporcionar información acerca de uno o varios de los elementos siguientes:
1. Relativa a un proceso o estado fisiológico o patológico. 2. Relativa a deficiencias físicas o mentales congénitas. 3. Relativa a la predisposición a una dolencia o enfermedad. 4. Para determinarla seguridad y compatibilidad con posibles receptores. 5. Para predecir la respuesta o reacción a un tratamiento. 6. Para establecer o supervisar medidas terapéuticas. Los recipientes para muestras se consideran también productos sanitarios para diagnóstico in vitro. |
| Producto sanitario legacy | Cualquier producto sanitario que cumplía con la normativa previa al EU-MDR, pero que aún no está certificado por la normativa actual. |
| Regulation (EU) 2017/745 en MDR | Reglamento europeo de productos sanitarios. |
| Regulation (EU) 2017/746 en IVDR | Reglamento europeo de productos in vitro. |
| Marcado CE | Marcado por el que un fabricante indica que un producto es conforme con los requisitos aplicables establecidos en el presente reglamento y en otra legislación de armonización de la Unión aplicable que prevea su colocación (MDR, artículo 2.43). |
|
CE: Certificación Europea; EU: Unión Europea; IVDR: regulación de dispositivos in vitro; MDR: reglamento de productos sanitarios. |
|
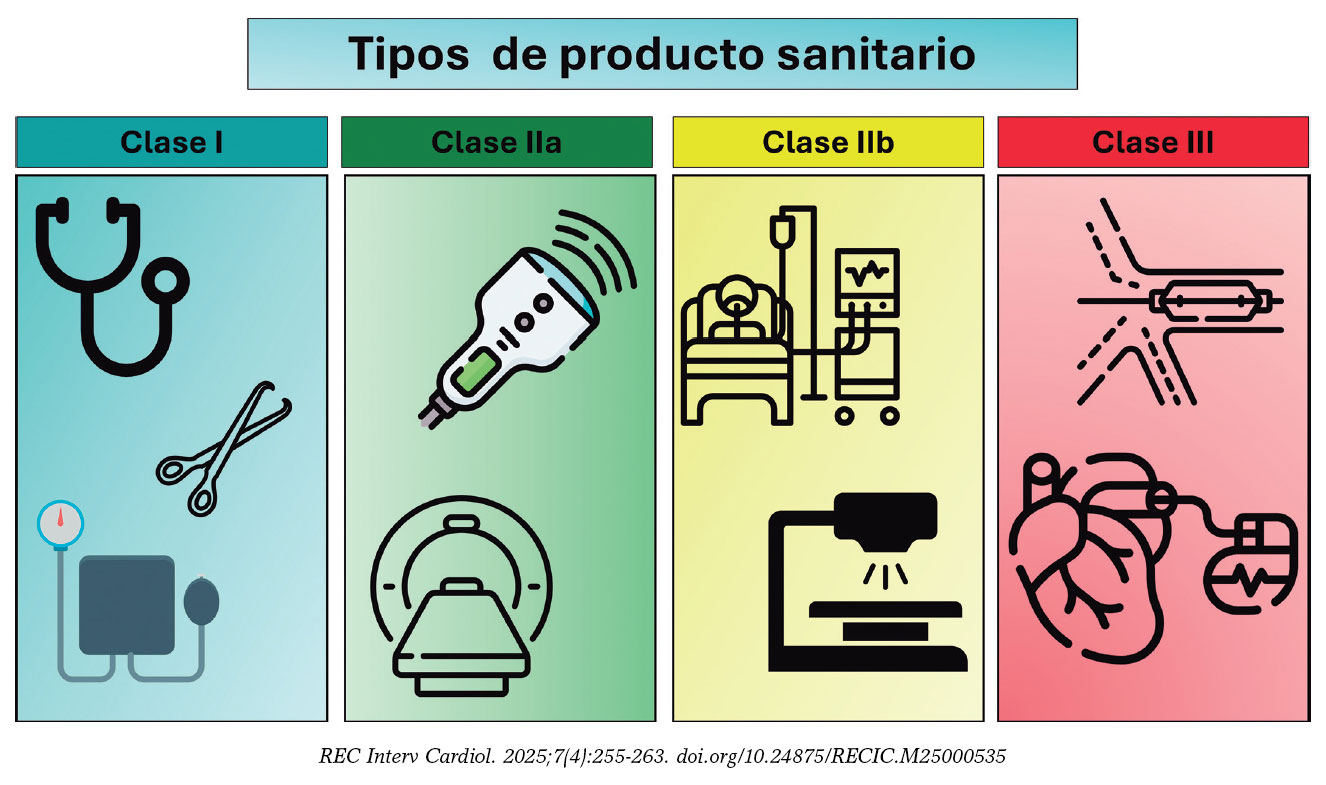
Figura 2. Clasificación de los distintos productos sanitarios y ejemplos de ellos. Los iconos han sido realizados por Kiranshastry, Vectorslab, Iconsmeet, Freepik y N.style en www.flaticon.com.
- – Clase I: productos de bajo riesgo para el paciente, generalmente no invasivos. Ejemplos: estetoscopio, electrodos de electrocardiograma, instrumental quirúrgico reutilizable y esfigmomanómetro no automatizado. Las clases específicas son Is (estéril), Im (función de medición) e Ir (reutilizable).
- – Clase IIa: productos de riesgo moderado que se utilizan de manera invasiva, pero temporalmente. Ejemplos: aguja hipodérmica, estetoscopio electrónico, equipos electrónicos de medición de la presión arterial, electrocardiógrafo, máquina de ultrasonido de diagnóstico (como ecocardiografía) y sistema de resonancia magnética.
- – Clase IIb: productos de mayor riesgo que pueden tener contacto prolongado con el cuerpo o que controlan funciones vitales. Ejemplos: bombas de infusión, dispositivos de monitorización y alarma en cuidados intensivos, ventiladores, desfibriladores externos y dispositivos de diagnóstico que emiten radiación ionizante.
- – Clase III: productos de alto riesgo, en general implantables y que tienen un impacto significativo en la salud del paciente. Ejemplos: catéteres y guías cardiovasculares, stents metálicos y liberadores de fármacos, válvulas cardiacas protésicas y dispositivos de reparación de válvulas, dispositivos implantables para otras intervenciones estructurales cardiovasculares, electrodos de electrofisiología y catéteres y equipos de ablación, dispositivos electrónicos implantables cardiovasculares incluidos marcapasos, desfibriladores automáticos implantables y terapia de resincronización cardiaca, sistemas de circulación extracorpórea, oxigenador extracorpóreo de membrana, bomba de balón intraaórtico y dispositivos de asistencia ventricular izquierda.
PERSPECTIVA DE LAS AGENCIAS REGULADORAS, LOS ORGANISMOS NOTIFICADOS Y LOS COMITÉS ÉTICOS
Para la obtención del marcado CE, un PS debe cumplir una serie de requisitos impuestos por los distintos organismos reguladores. Tales requisitos dependen de la finalidad para la que se presenta y la clasificación de riesgo de cada tipo de PS. Por otra parte, en cuanto a la realización de investigaciones clínicas con PS cubiertas por el reglamento 2017/745, en la tabla 2 se indican ciertos requisitos fundamentales. A continuación, se comentan las particularidades de todos los organismos implicados.
Tabla 2. Requisitos para la realización de investigaciones clínicas con productos sanitarios
| Investigaciones con productos sanitarios | CEIm | AEMPS | Conformidad de dirección | Seguro | Artículo |
|---|---|---|---|---|---|
| Sin CE | Sí | Autorización | Sí | Sí | Artículo 62 (objetivo: evaluar conformidad CE) |
| Con CE, distinta finalidad | Sí | Autorización | Sí | Sí | Artículo 74, Apartado 2 |
| Con CE, misma finalidad y siguiendo las instrucciones de uso | |||||
| Con procedimientos adicionales invasivos o gravosos | Sí | Notificar | Sí | No | Artículo 74, Apartado 1 |
| Artículo 73 (notificar NEOPS/EUDAMED) | |||||
| Sin procedimientos adicionales invasivos ni gravosos | Sí | No | Sí | No | |
|
AEMPS: Agencia Española de Medicamentos y Productos Sanitarios; CE: Certificación Europea; CEIm: comité de ética en investigación médica. |
|||||
Comité de ética de la investigación con medicamentos
Obtener la aprobación por el CEIm es el primer paso que toda investigación debe superar para su realización. Debe distinguirse entre el comité de ética de la investigación, que regula proyectos de investigación biomédica en la Ley 14/2007, y el CEIm8, que además está acreditado para emitir un dictamen en estudios clínicos con medicamentos y PS, regulado por el Real Decreto 1090/20159. No se han publicado aún los criterios que un CEIm debe cumplir para la evaluación de investigaciones clínicas con PS. Los estudios de funcionamiento de PS para diagnóstico in vitro no requerirán evaluación por un CEIm; en algunos casos, puede ser por un comité de ética de la investigación según el EU-IVDR10. La función de todo CEIm es salvaguardar los derechos, la seguridad y el bienestar de los sujetos participantes. Para este fin, en su composición debe contar con profesionales sanitarios, vocales ajenos a las profesiones sanitarias (incluyendo graduados en derecho), representantes de pacientes y un experto en el Reglamento General de Protección de Datos; adicionalmente, puede solicitar el asesoramiento de expertos cuando no reúna los conocimientos o la experiencia necesarios para evaluar un estudio (como puede ser en la investigación de procedimientos quirúrgicos, técnicas diagnósticas o terapias avanzadas). Consultar a expertos en materia de PS es muy frecuente, dadas su variedad y diferente finalidad y naturaleza que, en ocasiones, hacen difícil elucidar si un dispositivo se trata de un PS (por ejemplo, en el caso de aplicaciones informáticas). La evaluación del CEIm requerirá:
- Evidencia clínica previa de que el PS ejercerá su función y tendrá garantías de seguridad.
- Metodología adecuada para dar respuesta a los objetivos, definiendo claramente en el protocolo la justificación, la población a estudio, el tamaño muestral, las variables a incluir, el proceso de reclutamiento y aleatorización, etc.
- Desde el punto de vista ético, debe garantizar el cumplimiento de los principios éticos, especialmente en relación con la obtención del consentimiento informado, y cumplir los principios de la Declaración de Helsinki11 y la norma internacional ISO 14155:2021 («Investigación clínica de productos sanitarios para humanos. Buenas prácticas clínicas»)12.
Según el Real Decreto 192/2023, para la investigación en PS se exigirá la conformidad de la dirección del centro o de los centros donde vaya a realizarse, así como un dictamen favorable del CEIm local o, en caso de tratarse de un estudio multicéntrico, un dictamen emitido por un CEIm del territorio nacional único y vinculante3. En cualquier caso, debe considerarse que cada centro tiene que gestionar aspectos administrativos internos para permitir la realización del estudio. El plazo para que cada CEIm emita su dictamen no se especifica en el Real Decreto 192/2023, por lo que depende en gran medida de las características de cada comité.
Considerando la evidente complejidad de la nueva regulación, representantes de distintos CEIm en la Asociación Nacional de Comités de Ética de la Investigación (ANCEI), junto con la AEMPS, acordaron en 2023 formar un grupo de trabajo para armonizar la evaluación de las investigaciones en PS. Se espera que de esta colaboración surja en los próximos meses un memorando en conjunto de instrucciones para facilitar el proceso de certificación y armonizar la documentación requerida por ambas entidades; por ejemplo, el memorando definirá los plazos de los que se dispondrá para emitir un dictamen.
Agencia Española de Medicamentos y Productos Sanitarios
En España, la AEMPS es la autoridad competente encargada de la aplicación y la supervisión del reglamento EU 2017/745. La AEMPS ha elaborado una serie de documentos y manuales con el objetivo de aclarar el proceso, los requisitos y la documentación que es necesario presentar para su evaluación de estudios con PS13. En la tabla 2 se presenta un resumen de los requerimientos para la realización de investigaciones clínicas con PS.
La AEMPS sigue trabajando en revisar nuevos documentos. Quedan pendientes los relativos a PS para diagnóstico in vitro y el documento conjunto de los CEIm con la AEMPS ya mencionado. Además, desde el Medical Device Coordination Group, que es el grupo europeo implicado en la regulación de los PS, también se publican guías cuyo cumplimiento es de carácter voluntario, pero que reflejan la actualidad y el consenso en estos temas desde el punto de vista europeo14. Respecto a los estudios del funcionamiento de PS para diagnóstico in vitro, sus requerimientos, en muchos casos, no precisarán autorización ni notificación por la AEMPS, y además podrían ser evaluados por un comité de ética de la investigación. Sin embargo, aquellos estudios de funcionamiento de PS para diagnóstico in vitro que pudieran ser gravosos para la salud o presentar un riesgo para los pacientes se regulan por el artículo 58 de su propia regulación (EU-IVDR 2017/74610), y sus requerimientos son similares a los mostrados en la tabla 2. Entre los procedimientos de riesgo se incluyen la toma de muestra quirúrgicamente invasiva únicamente a efectos del estudio, los procedimientos invasivos adicionales y otros riesgos para los sujetos participantes (como procedimientos intervencionistas o pruebas diagnósticas que puedan influir en las decisiones clínicas).
Existen también estudios combinados que implican a la vez medicamentos y PS, a los que se aplicarán ambas regulaciones. Un caso que conviene clarificar es el de los productos integrados. Los estudios con estos productos no serían estudios combinados, ya que no implican medicamentos y PS. Un producto integrado es un PS o un medicamento, y se le aplicaría solo la legislación correspondiente. El criterio que debe seguirse es fundamentalmente su mecanismo de acción principal: si es el de un medicamento o el de un PS. Un ejemplo de producto integrado que es un PS sería un stent farmacoactivo, cuyo mecanismo de acción principal es el efecto mecánico para dilatar la arteria coronaria y en el que la pequeña cantidad de sustancia medicinal que incorpora tendría una acción antiproliferativa accesoria. Por otra parte, una jeringa precargada de insulina también se trataría de un producto integrado, pero en este caso el mecanismo de acción principal es el del medicamento (insulina). La «parte del producto» que sería PS por separado actuaría únicamente como sistema de administración del medicamento.
El procedimiento de autorización de investigaciones clínicas con PS por la AEMPS, así como otra información relativa a la regulación de PS, se encuentran recogidos en las instrucciones publicadas en su página web, que en la actualidad están siendo revisadas13. El procedimiento es, de forma muy resumida: 15 días de plazo para la validación de la documentación, 20 días para que el promotor responda a los posibles requerimientos de la AEMPS y 10 días para la evaluación de las respuestas por la AEMPS. Una vez admitida a trámite la solicitud, comienza el periodo de evaluación de la investigación clínica, que será de 45 días, aunque la AEMPS puede disponer de 20 días naturales más con el fin de consultar a expertos. La AEMPS podrá presentar objeciones al promotor, así como requerir información complementaria, quedando en este caso suspendidos los plazos y disponiendo el promotor de 10 días hábiles (ampliables a 5 más) para responder. Es importante mencionar que la AEMPS requerirá la aprobación del CEIm, por lo que se recomienda enviar la documentación con el dictamen del CEIm ya emitido o en proceso avanzado; de no ser así, la AEMPS denegará automáticamente la investigación cuando se cumpla el plazo. Una vez finalizada la investigación clínica, en 15 días naturales el promotor lo notificará a la AEMPS. Si se realiza una paralización temporal o una finalización anticipada, el promotor deberá notificarlo en 15 días naturales y proporcionará una justificación; si se debe a motivos de seguridad, deberá informar a la AEMPS en un plazo de 24 horas. Para el informe final de la investigación, el promotor dispondrá de 1 año desde la finalización de la investigación o de 3 meses si esta ha sido finalizada de manera anticipada. Se debe destacar que en el artículo 69 del EU-MDR se especifican las condiciones en las que las investigaciones deben disponer de un seguro o garantía financiera para los sujetos de investigación1. Como se indica en la tabla 2, las investigaciones con PS sin marcado CE, o con marcado CE, pero utilizadas con una finalidad distinta de la aprobada cuando obtuvieron la CE, deberán exigir un seguro para los sujetos.
Un pilar fundamental para el desarrollo de la EU-MDR es la base de datos EUDAMED (European Database on Medical Devices, cuyo objetivo es mejorar la transparencia pública, facilitar la trazabilidad de dispositivos, facilitar la supervisión del mercado y reforzar la coordinación entre los Estados miembros. Esta base de datos, que será accesible para las organizaciones estatales, aún no se encuentra disponible en el momento de la publicación de este documento. Por lo tanto, la AEMPS sigue utilizando su registro general para la tramitación de las autorizaciones de investigaciones clínicas con PS y de los estudios del funcionamiento con PS para diagnóstico in vitro. Para los casos que únicamente requieren notificación (tabla 2) se está utilizando la base de datos denominada NEOPS (notificación de investigaciones clínicas con productos sanitarios), hasta que EUDAMED se implemente de manera definitiva.
Organismo notificado
Un ON es una organización designada por un Estado miembro de la Unión Europea (u otros países en el marco de acuerdos específicos) para evaluar la conformidad de determinados productos antes de introducirlos en el mercado. El único ON creado en España es el Centro Nacional de Certificación de Productos Sanitarios, que dispone de competencia y capacidad para designar y realizar el seguimiento de PS. El ON exige documentación técnica normalizada y que cumpla con los requisitos necesarios de nivel de evidencia previo y conformidad con la regulación general de seguridad y funcionamiento. En su página web se encuentran los documentos necesarios para realizar cada uno de los trámites y guías orientativas (tabla 1 del material adicional).
Desde el comienzo de su actividad, el 15 de septiembre de 2022, el ON español ha sido requerido por 142 empresas solicitantes, con las cuales se han llegado a firmar 100 acuerdos. Estos acuerdos se definen como un contrato formal entre el ON y el fabricante que permite al ON realizar una evaluación del PS, definiendo el alcance de la evaluación, las obligaciones del fabricante y del ON, las condiciones para mantener, renovar o suspender la CE, y aspectos de confidencialidad y responsabilidad. Entre estos acuerdos, se han emitido 40 certificados que incluyen en torno a 300 productos (29 certificados de calidad y 11 de documentación técnica [PS de clase IIb implantables o de clase III]). El tiempo habitual entre el registro del PS y la firma del acuerdo es de 1 a 4 semanas. No obstante, en ocasiones este trámite puede alargarse más de 2 meses debido a una serie de dificultades que el ON presenta, como son:
- – Importante carga de trabajo: actualmente el ON tiene 275 productos por revisar, de los cuales el 25% son implantables, que requieren una evaluación más exhaustiva y exigente.
- – Estructura de la documentación técnica no adaptada: a pesar de los esfuerzos por generar una documentación estandarizada, habitualmente se requieren múltiples correcciones de la documentación, lo que retrasa el proceso de certificación.
- – Consulta a expertos: ciertos PS requieren una evaluación por un panel de expertos. De hecho, el artículo 56 declara que para los PS de clase IIb o III es obligatoria la consulta con un panel de expertos1. Encontrar expertos en determinados PS es en ocasiones una tarea difícil, teniendo en cuenta que algunos de los PS son para aplicaciones muy específicas, por lo que este hecho puede retrasar aún más la evaluación.
Es de especial importancia considerar que existe una fecha límite para la certificación de los PS con una certificación previa. Los PS que no hayan obtenido la CE bajo la nueva normativa para este plazo ya no podrán comercializarse, y podrían verse obligados a ser retirados del mercado. Este límite ya ha sido aplazado por el riesgo de escasez de productos en el mercado, la limitación de los ON y la influencia de la pandemia de COVID-19, siendo las fechas actuales el 31 de diciembre de 2027 para los PS de clase III y IIb implantables y el 31 de diciembre de 2028 para los de clase IIb no implantables, IIa, Is (estériles) e Ir (función de medición)15. Sin embargo, aunque estas fechas pueden parecer lejanas, el Centro Nacional de Certificación de Productos Sanitarios invita a los fabricantes a realizar el procedimiento de certificación sin demora.
Una limitación importante de los ON es la ausencia de un diálogo previo con los fabricantes. Este hecho puede dificultar el desarrollo de estudios clínicos adecuadamente diseñados que, en ocasiones, serían considerados inadecuados por el ON tras ser completados. Trabajar en conjunto en el diseño de los estudios clínicos podría ser beneficioso para ambas partes, reduciendo el tiempo necesario para la aprobación del PS y facilitando la realización del estudio y su revisión.
La documentación requerida es compleja y supone multitud de dudas y preguntas. Por ello, se han facilitado varios enlaces de interés a documentos y páginas web oficiales de los organismos correspondientes, donde estos aspectos complejos y concretos se explican con detenimiento (tabla 1 del material adicional).
PERSPECTIVA DE LA INDUSTRIA Y DE LOS FABRICANTES
Amenazas y retos
La industria tecnológica ha requerido una serie de profundas adaptaciones a la nueva normativa, implicando esta multitud de amenazas y dificultades. Según la asociación europea COCIR (European Trade Association representing the medical imaging, radiotherapy, health ICT and electromedical industries), la nueva regulación ha incrementado significativamente el tiempo hasta la certificación, aumentando un 157% el tiempo hasta la preparación de los documentos y un 263% hasta su aprobación final. También se ha elevado un 262% el coste y ha habido que incrementar un 5-9% el personal dedicado a la regulación, implicando una pérdida de profesionales y de presupuesto en el sector de investigación, desarrollo e innovación16. Una encuesta dirigida a diferentes empresas fabricantes en la industria de PS, realizada por MedTech Europe, encontró que los principales retos identificados son la insuficiente evidencia clínica generada mientras los dispositivos están en el mercado, la ausencia de una clara definición de lo que se considera información clínica suficiente y la dificultad para acordar con el ON un plan apropiado de seguimiento tras la comercialización17. Por otra parte, en una pregunta abierta con la que se evaluaron los principales obstáculos se mencionaron los costes asociados, los plazos, la divergencia de requerimientos en los distintos Estados y los importantes requisitos de las investigaciones clínicas. En este sentido, el Real World Data y el Real World Evidence son fuentes de información clínica relevantes. El Real World Data hace referencia a la información obtenida fuera de los ensayos clínicos tradicionales; puede extraerse de historias clínicas, registros de pacientes, bases de datos de seguros o estudios observacionales o de cohortes. El Real World Evidence implica el análisis o la interpretación del Real World Data, que se limita a datos crudos. De esta manera, el Real World Evidence permite generar evidencia sobre seguridad, efectividad, resultados de salud y uso de PS en la práctica diaria. No obstante, en la encuesta se observa que los ON aceptarían este tipo de evidencia mayormente como fuente adicional, que un 23% de los ON no la aceptarían en absoluto y que solo un 12% la aceptarían como única fuente de evidencia clínica. Entre el tipo de fuentes, los registros clínicos serían los más aceptados, quedando el resto de los formatos (encuestas, registros administrativos, etc.) muy por debajo en cuanto a aceptación.
Una amenaza particularmente sensible del desarrollo de PS afecta a aquellos destinados a pacientes vulnerables o con enfermedades huérfanas. Los PS cuyo ámbito de utilización sea en pacientes pediátricos o mujeres embarazadas corren el riesgo de no poder obtener la evidencia clínica suficiente para cumplir con los requerimientos necesarios para la obtención del marcado CE. De igual manera, si se exige una evidencia clínica sólida para un PS en enfermedades huérfanas, cuya prevalencia e incidencia son mínimas, existe el riesgo de que las empresas encargadas no sean capaces de cubrir los gastos de este desarrollo, ya de por sí complicado debido a la idiosincrasia de este tipo de enfermedades infrecuentes.
Competitividad con otros mercados
Muchas de estas limitaciones suponen una grave amenaza para PYME y start-up, que suelen presentar una estructura financiera menos sólida y consolidada que las grandes empresas. El incremento de los costes y la necesidad de personal adicional desincentiva el desarrollo de PS en Europa. Por lo tanto, algunas start-up, como Aortyx, han decidido enfocar el desarrollo de sus PS en sistemas más favorables, como el de Estados Unidos, cuyo principal atractivo desde el punto de vista burocrático está fundamentado en 2 puntos clave. El primero es la mayor rapidez de los trámites por parte de la Food and Drug Administration (FDA), que tiene un tiempo máximo de 90 días para dar una respuesta, mientras que los ON de Europa tardan una media de 50 días en simplemente aceptar el caso. En segundo lugar, resulta de especial interés la posibilidad de mantener un diálogo con la FDA para establecer qué tipo de estudio clínico sería el más adecuado para obtener su aprobación, en reuniones no vinculantes a través del programa Q-sub. En Europa, queda en manos del fabricante el diseño del estudio, sin posibilidad de recibir retroalimentación por parte del organismo regulador antes de finalizarlo, arriesgándose así a obtener una valoración desfavorable. En consecuencia, se vería obligado a repetir el estudio, con la sustancial inversión que esto implica. Aun así, en el artículo 57 de la EU-MDR se recomienda a los fabricantes consultar de manera voluntaria a un panel de expertos sobre la estrategia de desarrollo clínico y propuestas de investigación1. Además, en los Estados Unidos, algunos programas, como el Breakthrough Device Designation, pueden acelerar aún más el proceso priorizando aquellos PS novedosos considerados disruptivos y que traten enfermedades graves, que con frecuencia son enfermedades cardiovasculares18.
Aunque los anteriores motivos pueden considerarse suficientes para la priorización hacia Estados Unidos de ciertas empresas fabricantes de PS, la rentabilidad financiera también tiene una gran relevancia. El mercado americano ofrece, desde esta perspectiva, una serie de ventajas frente a la Unión Europea. La primera es la mayor morbilidad de la población estadounidense, y la segunda es el significativo mayor precio con que la mayoría de los PS se comercializan. En conjunto, todo esto hace que no solo desde el punto de vista burocrático, sino también desde el puramente financiero, el acceso al mercado sea más prometedor para la mayoría de pequeñas empresas tecnológicas sanitarias. De igual manera, y por razones similares, otros mercados como China y Japón resultan incrementalmente atractivos para las empresas y suponen una amenaza de reenfoque de los esfuerzos regulatorios y, en consecuencia, de desabastecimiento de PS innovadores en el mercado europeo.
Ejemplo de un estudio tras la comercialización
Con el objetivo de obtener el marcado CE MDR para un PS, la empresa iVascular realizó un plan de desarrollo clínico para un producto coronario de clase III legacy, un catéter-balón de angioplastia coronaria, tras su comercialización. Los PS legacy son aquellos certificados por la normativa previa y que disponen de un periodo para cumplir con la normativa actual. El seguimiento tras la comercialización debe confirmar la seguridad y el funcionamiento del producto a lo largo de su vida útil prevista, identificar y analizar riesgos emergentes, garantizar la aceptabilidad beneficio-riesgo e identificar posibles usos sistemáticos indebidos o no contemplados. Inicialmente se preparó el plan de desarrollo clínico en el que se establecieron 2 actividades. La primera fue una actividad formulario en la que se realizó una encuesta específica sobre el producto incluyendo variables de seguridad y funcionamiento clínico, y se envió a cardiólogos intervencionistas usuarios del dispositivo. La segunda actividad consistió en una investigación clínica, observacional, prospectiva y multicéntrica, con el objetivo de evaluar la seguridad y el funcionamiento clínico en población del mundo real y siguiendo las indicaciones de uso del fabricante. El estudio fue aprobado por los CEIm de cada centro. Tras completar el tamaño de la muestra y realizar el análisis estadístico, se elaboró un informe clínico que fue enviado al ON, donde se validó el resultado del PS y se otorgó el marcado CE.
A pesar del éxito de esta empresa, en su experiencia reconoce que la falta de retroalimentación por parte del ON, cuyo asesoramiento y aprobación podrían facilitar la elaboración del plan de desarrollo clínico, y la ausencia de una plantilla para el plan de desarrollo clínico y para la actividad formulario, supusieron limitaciones significativas en el procedimiento. Un proceso similar realizado en los Estados Unidos habría podido beneficiarse de un diálogo con la FDA para definir el tipo de estudio de manera consensuada e incluso aprovechar el programa Breakthrough Device Designation para acelerar el proceso. En China y Japón, aunque los plazos están más definidos, también se requiere un estudio clínico local para la aprobación del dispositivo. A pesar de estas ventajas, cabe señalar que disponer de un diálogo no garantiza una aprobación más acelerada, pues dependerá de las exigencias del estudio clínico requerido.
PERSPECTIVA DE LOS COLABORADORES
Los exigentes requerimientos para el desarrollo de un PS han llevado a que la mayoría de los investigadores y de los fabricantes necesiten el apoyo de empresas colaboradoras para el correcto cumplimiento de las distintas necesidades de los cada vez más complejos estudios clínicos. Existen una serie de aspectos en los que las empresas colaboradoras pueden ofrecer un servicio en el desarrollo de PS. De hecho, por las limitaciones existentes para los investigadores médicos y desarrolladores de PS existen las CRO (Contract Research Organization), empresas que ofrecen apoyo a la investigación clínica desde un enfoque global, gestionando el diseño del ensayo clínico, la recogida y el análisis de los datos, las cuestiones regulatorias, la monitorización del estudio clínicos y las auditorías y el control de calidad. A continuación, se mencionan las actividades para las que los investigadores y fabricantes demandan apoyo de colaboradores externos.
Elaboración y análisis de bases de datos
La creación de bases de datos adecuadas supone una parte crucial en el desarrollo de investigación clínica para garantizar el registro de todas las variables importantes que influyen en el estudio. Además, se debe garantizar que estas bases de datos sean accesibles a los investigadores y que se completen de manera adecuada. Para este fin, las empresas que dan soporte al diseño de bases de datos pueden ser un refuerzo esencial para los investigadores en la creación de estas y de los formularios adecuados, el seguimiento clínico, la curación y el análisis de los datos. Estas habilidades pueden requerir conocimientos específicos de informática de los que los investigadores no siempre disponen. Adicionalmente, la disponibilidad de bases de datos conjuntas en línea permite minimizar el riesgo de errores de transcripción y de errores en el seguimiento clínico mediante la adecuada coordinación de múltiples centros en estudios o ensayos clínicos multicéntricos. La externalización de este proceso puede aliviar la carga de trabajo de los investigadores y evitar errores en cada uno de los pasos.
Unificación de registros sanitarios
Una gran barrera en España para la realización de estudios de Real World Evidence es la limitación al acceso nacional de los datos. El hecho de que cada comunidad autónoma disponga de bases de datos independientes no accesibles a todos los investigadores nacionales dificulta la extracción y el análisis de datos a escala global. En este sentido, se han elaborado plataformas específicas con el objetivo de unificar bases de datos sanitarias en regiones con bases de datos heterogéneas, como ocurre en España. Una plataforma unificada podría aportar una serie de beneficios respecto al sistema actual. Para empezar, desde el punto de vista del paciente individual, que podría desplazarse por el territorio nacional y disponer de los resultados de pruebas previas realizadas en otras comunidades autónomas, facilitaría la asistencia sanitaria. Por otro lado, en cuanto a la investigación clínica, permitiría la extracción de datos unificados, aportando versatilidad en la exportación y comunicación con organizaciones y Estados, y la participación en proyectos de investigación conjuntos.
Consideraciones éticas y legales
Mientras que disponer de una gran cantidad de datos sanitarios puede ser una idea atractiva para los investigadores, este hecho conlleva cuestiones éticas sobre la protección de datos relativa a los pacientes. Existen asesorías especializadas en la seguridad de la información en el sector sanitario. Esta destaca por la compleja legislación existente en el ámbito de la seguridad del paciente y de la privacidad como derecho fundamental de los individuos. Es habitual que el investigador o el fabricante que debe centrar su capacidad de trabajo e intelectual en el desarrollo de una investigación o de un PS desconozca todos los matices de la compleja legislación en este ámbito concreto. Por ello, este tipo de empresas pueden ser un apoyo sustancial en el desarrollo de la investigación con PS, asegurando el cumplimiento de todos aquellos requerimientos legales necesarios y facilitando la realización de otras tareas a investigadores y fabricantes.
PERSPECTIVA DEL INVESTIGADOR
Desde el punto de vista del investigador, la aparición de nuevas regulaciones implica un esfuerzo adicional para su estudio y evaluación con el fin de llevar a cabo los proyectos de una manera eficiente y efectiva. La nueva normativa ha generado multitud de dudas en la comunidad investigadora y ha enlentecido, al menos temporalmente, el desarrollo de PS. En este sentido, la organización de reuniones de actualización con un enfoque transversal facilita la comprensión de estas complejas normativas, apoyan al investigador y permiten establecer valiosas redes profesionales que potencian la colaboración científica, reguladora e industrial.
El papel del investigador en tanto que experto en determinados PS, como se ha visto a lo largo del documento, es también fundamental. Tanto la AEMPS como los CEIm requerirán con frecuencia la valoración de determinados proyectos por paneles de expertos, y a su vez se recomienda a los fabricantes consultar con expertos los proyectos antes de presentarlos a los organismos oficiales. Por lo tanto, disponer de un grupo de expertos sólido y con experiencia supone una necesidad crucial para llevar a cabo investigaciones eficaces y para valorar con criterio las nuevas solicitudes de PS en colaboración con los organismos oficiales.
Por último, es habitual que muchos proyectos interesantes, iniciados por el investigador para responder a una pregunta académica o clínica, se vean enlentecidos o incluso detenidos debido a la imposibilidad de conseguir fondos suficientes. Este aspecto es en especial importante en el desarrollo de ensayos clínicos aleatorizados, que son el paradigma de la investigación clínica, pues requerirán un coste mayor para cubrir la necesidad de seguro sanitario para los participantes. La investigación en PS, por otra parte, puede suscitar un enfoque menos atractivo al investigador. No obstante, no se debe menospreciar la importancia de los estudios posautorización, ya que gracias a este tipo de estudios se han identificados PS y fármacos con efectos no identificados en los ensayos clínicos, en ocasiones perjudiciales para los pacientes, que han llevado a su retirada del mercado.
PERSPECTIVA DEL PACIENTE
En este complejo panorama es fácil olvidar que el objetivo final del desarrollo de PS es la mejora de la salud poblacional y de los pacientes, que son los receptores últimos de los PS. Por ello, su perspectiva es muy importante, y dar a conocer la complejidad del desarrollo de tecnología en salud y la multitud de requisitos necesarios para obtener PS seguros y eficaces resulta beneficioso tanto para los propios pacientes como para el resto de las partes. Limitar la participación de los pacientes a la firma del consentimiento informado minimiza su participación en el proceso y puede generar desconfianza hacia los investigadores, los fabricantes y los organismos reguladores. En cambio, explicar el proceso de desarrollo de PS de manera clara y sencilla puede acercar este complejo mundo a los pacientes. Desde los CEIm, de hecho, ya existe la exigencia de contar con representantes de pacientes dentro del comité. Otros ejemplos podrían ser la consulta directa a grupos de pacientes durante el diseño de los estudios, su participación en la definición de prioridades de investigación, el desarrollo de materiales informativos comprensibles para los participantes, y su colaboración en la interpretación y la difusión de los resultados. Estas formas de participación buscan fortalecer la transparencia, mejorar la relevancia de los estudios y fomentar la confianza en los procesos de investigación. Todos estos aspectos son un paso importante para fomentar la participación de pacientes en estudios clínicos y asegurar un seguimiento y un cumplimiento activo que, finalmente, repercutirán de manera positiva en la salud individual y poblacional.
CONCLUSIONES
La regulación europea EU-MDR ha traído cambios sustanciales al proceso de desarrollo de PS con el fin de unificar y asegurar un desarrollo eficaz y seguro de nuevos dispositivos médicos. Pese a que su implementación conlleva retos y amenazas para la investigación en PS en la Comunidad Europea, la colaboración de los múltiples actores implicados y compartir sus perspectivas ayudan a comprender el procedimiento en conjunto y facilitan un proceso más armónico y unificado.
FINANCIACIÓN
La Fundación Epic organizó la reunión y se hizo cargo de todos los gastos. El Instituto de Empresa (IE) cedió las instalaciones para la reunión Epic MDR25.
CONSIDERACIONES ÉTICAS
Este artículo es una revisión de la literatura previamente publicada y no involucra estudios originales con seres humanos ni animales por parte de los autores. Por lo tanto, no fue necesario obtener la aprobación de un comité de ética.
DECLARACIÓN SOBRE EL USO DE INTELIGENCIA ARTIFICIAL
Para la redacción de este documento no se utilizó ningún programa de inteligencia artificial.
CONTRIBUCIÓN DE LOS AUTORES
J. Zubiaur y A. Pérez de Prado redactaron la primera versión del texto. J.R. Rumoroso Cuevas, M.C. Rodríguez Mateos, G. Hernández, I. Alfonso Farnós, M. Aláez, S. Pich, J. Martorell, E. Paredes, A. Escario, P.P. Fernández Rivero, E. Jiménez-Carles y A. Pérez de Prado contribuyeron a la elaboración del contenido de donde se obtuvo la información para la redacción del documento. Todos los autores revisaron la versión final del artículo.
CONFLICTO DE INTERESES
A. Pérez de Prado es editor asociado de REC: Interventional Cardiology; se ha seguido el procedimiento editorial establecido en la revista para garantizar la gestión imparcial del manuscrito. J.R. Rumoroso Cuevas es director general de la Fundación Epic. I. Alfonso Farnós es presidenta de la ANCEI. M. Aláez es trabajadora en la FENIN. S. Pich es trabajadora en iVascular. J. Martorell es fundador y trabajador de Aortyx. E. Paredes es fundador y trabajador de pInvestiga. A. Escario es trabajador en Madrija. P.P. Fernández Rivero es trabajador en Start Up SL. E. Jiménez-Carles es director general y trabajador de Soinde Neuro SL.
MATERIAL ADICIONAL

BIBLIOGRAFÍA
1. Regulation - 2017/745 - EN - Medical Device Regulation - EUR-Lex. Disponible en: https://eur-lex.europa.eu/eli/reg/2017/745/oj/eng. Consultado 18 Ene 2025.
2. Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU (Text with EEA relevance). OJ L abr 5, 2017. Disponible en:http://data.europa.eu/eli/reg/2017/746/oj/eng. Consultado 25 Ene 2025.
3. Ministerio de Sanidad. Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios. Sec. 1, Real Decreto 192/2023 mar 22, 2023 p. 42678-42706. Disponible en: https://www.boe.es/eli/es/rd/2023/03/21/192. Consultado 25 Ene 2025.
4. Fraser AG, Byrne RA, Kautzner J, et al. Implementing the new European Regulations on medical devices —clinical responsibilities for evidence-based practice:a report from the Regulatory Affairs Committee of the European Society of Cardiology. Eur Heart J. 2020;41:2589-2596.
5. Spitzer E, de la Torre-Hernández JM, Guðmundsdóttir Ingibjörg J, et al. Uso de registros cardiovasculares en procesos regulatorios:perspectivas del Colaboratorio Cardiovascular EU-MDR. REC Interv Cardiol. 2024;6:213-223.
6. DocsRoom - European Commission. Disponible en: https://ec.europa.eu/docsroom/documents/10337/attachments/1/translations. Consultado 21 Ene 2025.
7. Biomed Alliance. Biomed Alliance response to JRC consultation on medical devices clinical investigation summary reporting requirements. 10 sep 2018. Disponible en: https://www.biomedeurope.org/wp-content/uploads/2018/12/Biomed_Alliance_Response_to_JRC_Consultation_10_Sep_2018-2.pdf. Consultado 25 Ene 2025.
8. Jefatura del Estado. Ley 14/2007, de 3 de julio, de Investigación biomédica. Sec. 1, Ley 14/2007 jul 4, 2007 p. 28826-28848. Disponible en: https://www.boe.es/eli/es/l/2007/07/03/14. Consultado 25 Ene 2025.
9. Ministerio de Sanidad, Servicios Sociales e Igualdad. Real Decreto 1090/2015, de 4 de diciembre, por el que se regulan los ensayos clínicos con medicamentos, los Comités de Ética de la Investigación con medicamentos y el Registro Español de Estudios Clínicos. Sec. 1, Real Decreto 1090/2015 dic 24, 2015 p. 121923-121964. Disponible en: https://www.boe.es/eli/es/rd/2015/12/04/1090. Consultado 25 Ene 2025.
10. Regulation - 2017/746 - EN - Medical Device Regulation - EUR-Lex. Disponible en: https://eur-lex.europa.eu/eli/reg/2017/746/oj/eng. Consultado 17 Ene 2025.
11. WMA - The World Medical Association - Declaración de Helsinki de la AMM - Principios éticos para las investigaciones médicas con participantes humanos. Disponible en: https://www.wma.net/es/policies-post/declaracion-de-helsinki-de-la-amm-principios-eticos-para-las-investigaciones-medicas-en-seres-humanos/. Consultado 18 Ene 2025.
12. UNE-EN ISO 14155:2021 Investigación clínica de productos sanitarios. Disponible en: https://www.une.org/encuentra-tu-norma/busca-tu-norma/norma?c=N0066686. Consultado 18 Ene 2025.
13. Agencia Española de Medicamentos y Productos Sanitarios. 2023 Instrucciones de la AEMPS para la realización de investigaciones clínicas con productos sanitarios en España. Disponible en: https://www.aemps.gob.es/productos-sanitarios/investigacionclinica-productossanitarios/instrucciones-de-la-aemps-para-la-realizacion-de-investigaciones-clinicas-con-productos-sanitarios-en-espana/. Consultado 17 Ene 2025.
14. Guidance - MDCG endorsed documents and other guidance - European Commission. 2025. Disponible en: https://health.ec.europa.eu/medical-devices-sector/new-regulations/guidance-mdcg-endorsed-documents-and-other-guidance_en. Consultado 27 Ene 2025.
15. BOE.es - DOUE-L-2023-80401 Reglamento (UE) 2023/607 del Parlamento Europeo y del Consejo de 15 de marzo de 2023 por el que se modifican los Reglamentos (UE) 2017/745 y (UE) 2017/746 en lo que respecta a las disposiciones transitorias relativas a determinados productos sanitarios y a productos sanitarios para diagnóstico in vitro. Disponible en: https://www.boe.es/buscar/doc.php?id=DOUE-L-2023-80401. Consultado 23 Ene 2025.
16. COCIR. COCIR Position on the Future Governance Framework for the Medical Technologies Sector. Disponible en: https://www.cocir.org/position/cocir-position-on-the-future-governance-framework-for-the-medical-technologies-sector/. Consultado 18 Ene 2025.
17. MedTech Europe. MedTech Europe 2024 Regulatory Survey:key findings and insights. Disponible en: https://www.medtecheurope.org/resource-library/medtech-europe-2024-regulatory-survey-key-findings-and-insights/. Consultado 18 Ene 2025.
18. U.S. Food and Drug Administration. Breakthrough Devices Program. FDA. 11 de julio de 2024. Disponible en: https://www.fda.gov/medical-devices/how-study-and-market-your-device/breakthrough-devices-program. Consultado 18 Ene 2025.
