
RESUMEN
El 26 de mayo de 2021 entró en vigor el Reglamento Europeo de Productos Sanitarios (EU-MDR), que supuso un importante cambio en los requisitos de evaluación de los productos sanitarios en Europa. El EU-MDR Cardiovascular Collaboratory (EU-MCVC) se fundó con el fin de contribuir al desarrollo de vías más rápidas, eficientes y eficaces para la innovación de productos sanitarios cardiacos. Un registro es un sistema organizado que recoge datos uniformes y evalúa resultados específicos en una población definida por una enfermedad, afección o exposición. La mayoría de los registros se han desarrollado para mejorar la calidad de la atención y proporcionar información a médicos, hospitales y proveedores de servicios sanitarios. Los registros clínicos representan una construcción ideal para la colaboración científica, clínica y política. Describimos diversas experiencias de 5 países europeos y abordamos los componentes de calidad tradicionales en los ensayos clínicos. Se espera una colaboración continua entre académicos, especialistas en ensayos clínicos, representantes de pacientes, expertos en regulación, organizaciones de investigación, plataformas de registros, organismos reguladores y socios de la industria. La calidad de los datos es una preocupación primordial y los responsables de los registros deben optimizarla para cumplir con la normativa. Un enfoque colaborativo entre las partes interesadas en los dispositivos médicos puede mejorar la calidad de la atención, reducir los costes y proporcionar un acceso más rápido a tecnologías innovadoras, con el objetivo común de mejorar la atención y los resultados cardiovasculares.
Palabras clave: Ciencia reguladora. Registros clínicos. Ensayos clínicos.
ABSTRACT
On May 26, 2021, the European Medical Device Regulation (EU-MDR) entered into effect resulting in a major shift in the requirements for assessment of medical devices in Europe. The EU-MDR Cardiovascular Collaboratory (EU-MCVC) was founded to contribute to the development of faster, more efficient, and more effective pathways for innovation of cardiac medical devices. A registry is an organized system that collects uniform data and evaluates specified outcomes in a population defined by a disease, condition, or exposure. Most registries have been created to improve the quality of care and provide feedback to physicians, hospitals, and health providers. Clinical registries represent an ideal construct for scientific, clinical, and policy-making collaboration. We describe diverse experiences from 5 European countries and address the traditional quality components in clinical trials. Continued collaboration is expected among academics, clinical trialists, patient representatives, regulatory experts, research organizations, registry platforms, regulatory bodies, and industry partners. Data quality is a primary concern and registry leaders need to optimize data quality to become regulatory compliant. A collaborative approach among medical device stakeholders may improve quality of care, reduce costs, and provide faster access to innovative technologies, with the common objective of improving cardiovascular care and outcomes.
Keywords: Regulatory science. Clinical registries. Clinical trials.
Abbreviations EMA: European Medicines Agency. EU-MCVC: European Medical Device Regulation Cardiovascular Collaboratory. EU-MDR: European Medical Device Regulation. PMCF: Post-marketing clinical follow-up. RCT: Randomized controlled trial.
INTRODUCCIÓN
El 26 de mayo de 2021, se promulgó el Reglamento Europeo de Productos Sanitarios (EU-MDR) y la Unión Europea implementó un cambio importante en los requisitos en materia de investigación y desarrollo de dispositivos médicos1. No obstante, esta actualización legislativa coordinada permitió que cada país europeo adoptara este nuevo marco normativo aplicara su propio criterio, lo cual introdujo importantes curvas de aprendizaje. Los comités de ética, las autoridades competentes, los organismos notificados, las instituciones de salud académicas y no académicas, así como las organizaciones de investigación por contrato experimentaron retrasos, tiempos de espera más largos, una mayor carga de trabajo y, por ende, una pérdida de eficacia. Esto, a su vez, hizo que, en algunos casos, los fabricantes optaran por no priorizar a Europa como un lugar potencial para el desarrollo de nuevos tratamientos. Tres años después de la implementación del EU-MDR, se han superado las curvas de aprendizaje y Europa ha vuelto a ser una prioridad. No obstante, los nuevos requisitos y los mayores costes asociados abogan por la consecución de vías alternativas para generar datos en materia regulatoria.
Una actualización pertinente de este nuevo marco normativo es la necesidad de que los fabricantes lleven a cabo actividades de seguimiento clínico poscomercialización (PMCF) basadas en la recopilación de datos clínicos sobre el uso de dispositivos ya disponibles en el mercado. El propósito refleja el deseo de confirmar los requisitos de seguridad y rendimiento en condiciones normales de uso previsto del dispositivo, valorar los efectos adversos potencialmente raros y garantizar que la ratio riesgo-beneficio, específica de cada dispositivo, siga siendo favorable1. Aunque los estudios de poscomercialización eran algo habitual en el anterior marco regulador, el EU-MDR los ha convertido en obligatorios. Más allá de las consecuencias económicas, estos requisitos provocan, inevitablemente, una mayor carga de trabajo en aquellos hospitales en los que se emplean e implementan los dispositivos. Esta carga de trabajo adicional se podría aliviar estableciendo alianzas público-privadas para la recopilación y presentación de datos eficientes, efectivos y de alta calidad.
Un manejo exitoso de las enfermedades cardiacas requiere el uso o la implementación de dispositivos médicos; aquí el EU-MDR ha tenido un impacto esencial en el acceso a la investigación, la innovación y terapias mejoradas en la cardiología en Europa. En mayo de 2023, Cardialysis (Róterdam, Países Bajos) inició el EU-MDR Cardiovascular Collaboratory (EU-MCVC), una red internacional informal de expertos, a título voluntario y pro bono, que reúne a académicos europeos, ensayistas clínicos y expertos legisladores para colaborar con las partes interesadas en investigación clínica, tanto de ámbito local como internacional2. El propósito del EU-MCVC es crear una conversación dinámica y abierta para facilitar, en tiempo real, la implementación efectiva de la investigación clínica en Europa poniendo el énfasis en la navegación del EU-MDR. Los actores económicos en materia de investigación más importantes en esta colaboración son las organizaciones de investigación cardiovascular, las plataformas de registro, los organismos reguladores y los socios de la industria.
El abordaje prioritario de 2023 a 2024 es la definición, los requisitos y el establecimiento de registros clínicos cardiovasculares eficientes, efectivos y de alta calidad como una vía valiosa para la recopilación de datos PMCF. Este artículo aborda los siguientes 4 temas: a) la definición de registros y consideraciones relacionadas con el consentimiento informado; b) perspectivas de 5 líderes europeos sobre el establecimiento y desempeño de registros clínicos; c) la interacción entre los procesos de calidad de estudios clínicos tradicionales y los registros clínicos; d) los requisitos de datos de registro y su uso potencial y actual.
Este documento de perspectivas se redactó sobre la base de aportaciones voluntarias de todos los autores. El proceso de generación del manuscrito tuvo 2 componentes: a) un grupo de trabajo híbrido organizado por el EU-MCVC y Cardialysis, con miembros de la facultad que asistieron, sobre todo, en persona (11/13), el 8 de septiembre de 2023, en Róterdam (Países Bajos); y b) la compilación de presentaciones, debates y conclusiones, en un documento preliminar que fue revisado críticamente y ampliado por cada uno de los autores.
Definición de registros clínicos
La Agencia Europea de Medicamentos (EMA), U.S. Food and Drug Administration y el International Medical Device Regulators Forum son organismos que orientan en materia de definición de registros clínicos (tabla 1)3-6. Los elementos comunes de estas definiciones describen un registro como un sistema organizado que recopila datos uniformes y evalúa resultados específicos en una población definida por una enfermedad, patología o exposición. La definición que da el International Medical Device Regulators Forum se centra en la calidad asistencial dispensada al paciente, por lo que requiere un tamaño razonablemente extrapolable que sería más útil para informar a la hora de legislar. Sin embargo, la definición de la U.S. Food and Drug Administration adapta los objetivos a propósitos científicos, clínicos o políticos mientras que la definición de la EMA pone el acento en que la definición se haga a nivel del paciente, poniendo el foco del registro en la información sanitaria.
Tabla 1. Definición de registros clínicos
| Definición del International Medical Device Regulators Forum (IMDRF) |
| Un registro es un sistema organizado que, paralelamente a la práctica clínica diaria, recopila de manera continua y consistente datos relevantes, valora resultados significativos y da una cobertura integral a la población definida mediante la exposición a determinado/s dispositivo/s en una escala razonablemente extrapolable (como, por ejemplo, internacional, regional, sistema de salud), con el objetivo principal de mejorar la calidad asistencial del paciente |
| Definición de la U.S. Food and Drug Administration |
| Un registro es un sistema organizado que emplea métodos de estudio observacional para recopilar datos uniformes (clínicos y de otro tipo) para valorar los resultados específicos de una población definida por una enfermedad, patología o exposición determinada y que sirve uno o más propósitos científicos, clínicos o políticos predefinidos |
| Definición de de la Agencia Europea de Medicamentos (EMA) «Registro de pacientes» |
| Un sistema organizado que recopila datos uniformes (clínicos y de otro tipo) para identificar los resultados específicos de una población definida por una enfermedad, patología o exposición determinada. El término «paciente» resalta el enfoque del registro en la información de salud. Está ampliamente definido y puede incluir pacientes con una determinada enfermedad, mujeres embarazadas o lactantes o individuos con otras patologías, como defectos en el nacimiento o con características moleculares o genómicas |
| Elementos de una definición común de registro clínico del EU-MDR Cardiovascular Collaboratory |
| Sistema organizado |
| Recopila datos uniformes (de manera continua y consistente) (clínicos y de otro tipo) |
| Valora resultados especificados (significativos) |
| Población definida por una enfermedad, patología o exposición |
| Perspectivas del EU-MCVC sobre el tamaño de los registros clínicos |
| Registro clínico unicéntrico frente a multicéntrico |
| Registro clínico nacional integral (todos los centros) frente a no integral (centros seleccionados) |
| Registro clínico internacional integral (todos los centros) frente a no integral (centros seleccionados) |
| Redes de registros clínicos (varios registros fusionando bases de datos independientes a nivel del paciente o del registro) |
Cuando se habla de los requisitos del EU-MDR, una población se define por la exposición a un dispositivo específico, lo cual tiene importantes consecuencias para el establecimiento de plataformas de registro en Europa. La EMA define, al menos, 3 categorías de registros que, en circunstancias ideales, podrían estar interconectadas. En primer lugar, la EMA define los registros de enfermedades como aquellos registros de pacientes cuyos participantes están definidos por una enfermedad específica o características asociadas a la enfermedad, con independencia de su exposición a los tratamientos. Los registros de enfermedades son meramente observacionales. En segundo lugar, la EMA define los estudios basados en registros como la investigación de una cuestión en materia de investigación utilizando una población de pacientes dentro de un registro de pacientes. La interpretación del EU-MCVC es que esto alude a intervenciones de investigación o cuando la investigación clínica requiere intervenciones adicionales invasivas o complicadas o bien reglas de seguimiento. Lo ideal sería definir los análisis puramente observacionales o descriptivos bajo el paraguas de los registros de enfermedades. En tercer lugar, la EMA hace referencia a los registros de productos o dispositivos, que se suelen aplican a estudios PMCF. Los estudios PMCF deben cumplir la normativa que se aplica a los ensayos clínicos tradicionales (como, por ejemplo, a los estudios de un único grupo) en virtud del MDR, a menos que no se incorporen intervenciones adicionales invasivas o gravosas en el protocolo del registro.
El desarrollo de registros clínicos sostenibles podría mejorar la calidad asistencial dispensada, abaratar los costes y proporcionar un acceso más rápido a mejores tratamientos. La figura 1 ofrece un marco conceptual para la implementación de un registro clínico.
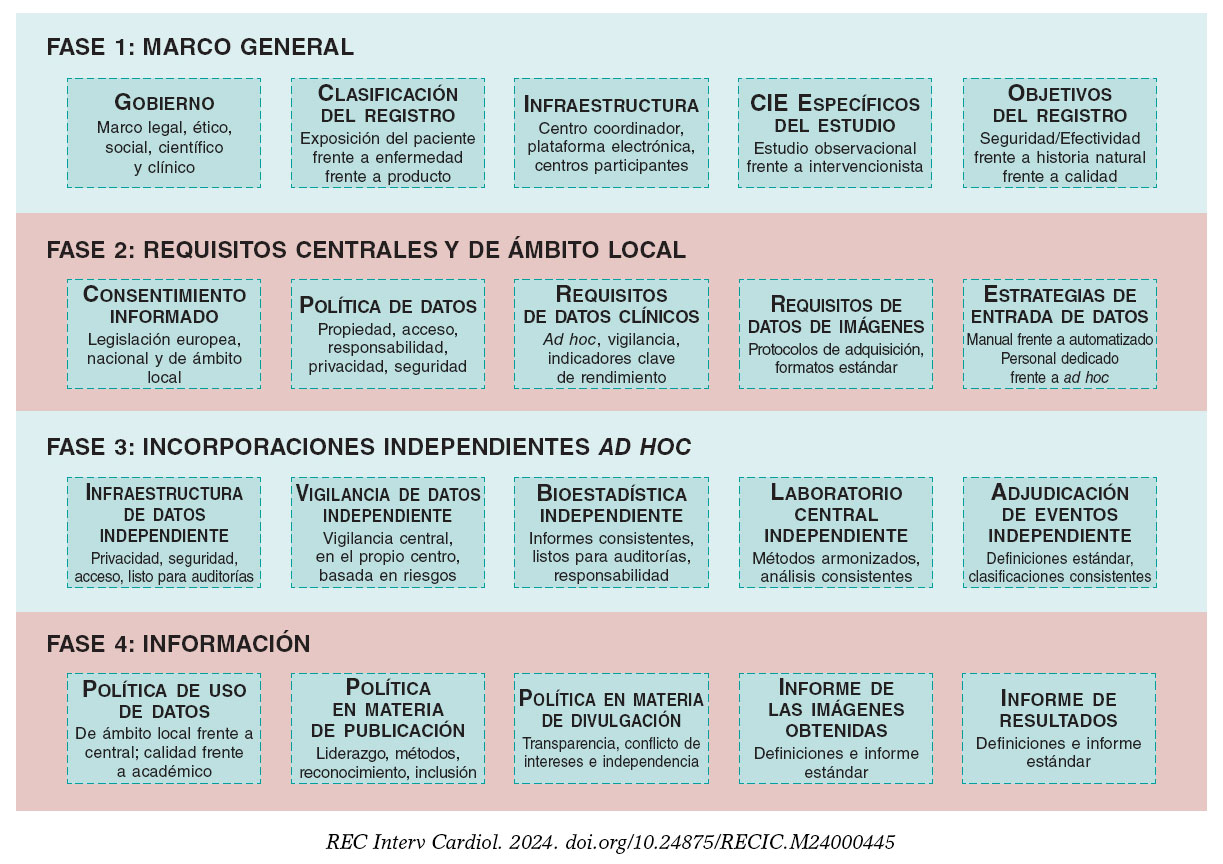
Figura 1. Marco conceptual para la implementación de un registro clínico. La fase 1 requiere fijar un marco legal y científico, así como también acordar y diseñar la distribución general de las tareas a realizar entre los colaboradores implicados. La fase 2 aborda el diseño e implementación del registro prestándose la mayor atención posible a los requisitos y calidad de los datos como común denominador. La fase 3 presenta actividades opcionales que pueden ser proporcionadas por partes independientes para aumentar la consistencia, calidad y confiabilidad a largo plazo. La fase 4 debe estar siempre lista y documentada cuando se esperan resultados. Todas las fases deberán ser debatidas e implementadas simultáneamente pues el producto final requiere la evaluación de estos 20 componentes. La documentación detallada, por escrito, de los acuerdos alcanzados debe obrar en poder del comité ejecutivo del registro. Todos los componentes de las fases 1, 2 y 4 son obligatorios y los de la fase 3, opcionales.
Antecedentes del utópico diseño de pacientes no seleccionados y de cómo los registros podrían ser la respuesta
Los ensayos controlados aleatorizados (ECA) son el patrón oro para valorar la seguridad y eficacia de las intervenciones médicas, ya que, por diseño, eliminan tantos factores de confusión como sea posible. Los ECA son el estándar para llevar a cabo una valoración previa precomercialización y suelen seleccionar a una población estrecha mediante criterios de elegibilidad cuidadosamente seleccionados, lo cual acarrea 4 consecuencias: a) se evitan la mayoría de los factores de confusión y se logran las estimaciones más puras posibles de los «efectos del tratamiento»; b) unos criterios de elegibilidad estrictos pueden dificultar la inscripción de pacientes, requiriendo más centros y más tiempo para completar la inscripción, si bien estos estudios suelen requerir tamaños de muestra más pequeños; c) como la población está altamente seleccionada, la validez externa limitada de la estimación del efecto de la intervención precisará estudios posteriores en poblaciones más grandes, normalmente en un contexto de poscomercialización; d) el diseño permite, solo, información limitada sobre posibles efectos adversos raros.
Como posible solución a los desafíos 2 y 3 mencionados arriba, se introdujo el «diseño de pacientes no seleccionados», que se caracteriza por tener criterios de elegibilidad simples. Este abordaje facilita la inscripción de una población de pacientes más representativa. No obstante, en un ensayo que evaluó stents coronarios7, quedaron sin inscribir tras el cribado, al menos, el 50% de los pacientes elegibles. Las principales razones fueron el proceso de consentimiento informado (el 33% no firmó el formulario de consentimiento informado, el 19% se negó a dar su consentimiento) y el 27% no cumplía los criterios de elegibilidad. Además, los pacientes que quedaron sin inscribir tuvieron peores resultados. Estas observaciones se han ido reproduciendo en muchas publicaciones posteriores. Otros posibles problemas a tener en cuenta en el abordaje de pacientes no seleccionados son: a) la incorporación de factores de confusión no controlados que podrían llevar a una “dilución” del efecto del tratamiento (inicialmente diseñado para una población específica y seleccionada) y a un efecto nulo en una comparativa aleatoria; b) los investigadores suelen excluir las presentaciones más graves como la insuficiencia cardiaca o el shock cardiogénico. En este sentido, el abordaje de pacientes no seleccionados sigue siendo selectivo. La llegada de investigaciones basadas en registros ofrece una oportunidad única de recopilar datos sobre todos los pacientes, sobre todo, en estudios meramente observacionales, y de comprender mejor los resultados en todas las subpoblaciones, especialmente aquellas tradicionalmente excluidas de los ensayos clínicos. La opinión del EU-MCVC es que las poblaciones no seleccionadas no deben tenerse en cuenta para realizar comparativas aleatorizadas tempranas a menos que se espere que un dispositivo beneficie a una población no seleccionada. En cambio, si se espera que el efecto de una intervención se dé, principalmente, en un subgrupo de pacientes, dichos subgrupos se deben investigar primero antes de emplear el abordaje de pacientes no seleccionados como abordaje inicial.
Consentimiento informado
Los pacientes ingresados o registrados en un centro sanitario no siempre son conscientes de que sus datos clínicos pueden ser utilizados de muchas formas. No obstante, por lo general, supondrán que la función más importante de los datos médicos, en lo que les atañe a ellos personalmente, es permitir que los profesionales sanitarios les den la mejor atención posible para mejorar su bienestar, calidad y esperanza de vida. No obstante, en función de cómo sea la legislación en materia de cuidados médicos, otros usuarios de sus datos pueden ser las compañías de seguros, las bases de datos nacionales, organizaciones asociadas (como, por ejemplo, las redes hospitalarias) y las bases de datos de calidad asistencial.
Cuando se invita a pacientes a participar en ensayos clínicos tradicionales, estos han de ser informados, en detalle, de los objetivos del estudio, los posibles riesgos y beneficios asociados, las cargas o compromisos adicionales, así como de cualquier otra información potencialmente relevante. De conformidad con el Reglamento general de protección de datos de la UE, los pacientes no solo ha de dar, voluntariamente, su consentimiento informado para inscribirse en una investigación clínica, sino que también deben dar, voluntariamente, su permiso para cada uso específico de sus datos; permiso que pueden retirar en cualquier momento1. A efectos de los ensayos clínicos y registros de pacientes, los datos de los participantes suelen estar codificados o seudonimizados, lo cual garantiza que sus identificadores personales no se compartirán fuera del centro médico que les trata. El uso de datos personales codificados se hace de conformidad con los requisitos de privacidad establecidos en el mencionado Reglamento general de protección de datos de la UE.
La cuestión de si se requiere consentimiento informado para ins- cribir participantes en un registro de pacientes depende de si la recopilación de datos del registro forma o no parte del estándar de cuidados (por ejemplo, el registro de calidad) y se define en los términos y condiciones del centro o en virtud de si el registro está fuera del alcance del estándar de cuidados. En el primer caso, el registro puede formar parte de los registros de las historias clínicas del paciente y las condiciones institucionales y regulatorias nacionales pueden, asimismo, no exigir el consentimiento específico para el registro. En el segundo caso, los pacientes deben dar su consentimiento antes de formar parte de un registro de pacientes. El EU-MCVC recomienda, siempre, coordinarse con el comité de ética local para definir la necesidad de consentimiento informado en el contexto de un registro clínico.
Lo que se espera de los registros de pacientes es que, solo, sean observacionales. En el caso de estudios de registros intervencionistas (es decir, con una intervención experimental) o estudios observacionales con complicadas intervenciones invasivas adicionales o reglas de seguimiento, se suele aceptar que los pacientes deben ser invitados a participar y que han de firmar un formulario de consentimiento informado. Los registros puramente observacionales también pueden precisar dicho proceso de consentimiento informado según sus objetivos y requisitos a nivel regional y nacional. Un formulario de consentimiento informado para registros observacionales debe indicar claramente que todos los datos codificados recopilados puedan emplearse para múltiples análisis de datos observacionales (de ámbito local o en la base de datos completa del registro) para evitar que el paciente tenga que volver a dar su consentimiento. Los pacientes siempre tienen el derecho a retirar su consentimiento informado.
Registros clínicos cardiovasculares: la experiencia española
España tiene una larga tradición de recopilación de datos basada en registros en cardiología intervencionista bajo el lema «la unión hace la fuerza» procedentes de una población de más de 47 millones de personas8. Al ser uno de los registros de calidad en cardiología intervencionista más antiguos que existen en la actualidad, la comunidad cardiológica ha podido proporcionar datos impactantes basados en registros. Desde 1990 lleva publicándose ininterrumpidamente un registro de ámbito nacional en cardiología intervencionista que ha ido recopilando datos a través de extensas encuestas realizadas cada año y que reflejan el volumen de actividad y no datos de pacientes9,10. En este sentido, en materia de investigación, el valor de este registro es muy limitado.
Los registros de pacientes empezaron como una colaboración académica entre colegas que voluntariamente y sin financiación externa desarrollaron bases de datos comunes para recopilar datos sobre intervencionismo cardiovascular y resultados clínicos durante el seguimiento. Todo empezó en 2004 y ha evolucionado desde los registros unicéntricos hasta una red académica interactiva multimodal. Estas aportaciones voluntarias fueron reflejo de innumerables horas de entrada de datos estructurados y planes de seguimiento. Un ejemplo destacado es el artículo seminal publicado en 2008 sobre trombosis del stent entre 23.500 pacientes de 20 centros españoles11. Otros registros realizados en años posteriores dieron lugar a más de 10 publicaciones. Esta red empezó con registros observacionales para luego incluir estudios aleatorizados y registros intervencionistas gracias a la cada vez mayor colaboración internacional.
Reconociendo una clara tendencia hacia la investigación colaborativa en un contexto de enfermedades complejas y abordajes terapéuticos complejos, la red multimodal española ha evolucionado hacia la Fundación Educación y Promoción de la Investigación en Enfermedades Cardiovasculares (EPIC). Fundada en 2016, en la actualidad EPIC está inmersa en tareas de investigación académica, investigación patrocinada por la industria y estudios observacionales y cuenta con un historial de 47 proyectos, incluidos 11 PMCF y vigilancia poscomercialización en virtud del MDR. En la actualidad, cada registro se configura como un estudio clínico, siguiendo las normas tradicionales ISO14155:2020 e incorpora el consentimiento informado de los pacientes. En la actualidad, EPIC está ampliando sus capacidades en investigación regulatoria en colaboración con socios locales e internacionales. Los registros se financian a través de subvenciones nacionales o de la propia industria. Como organización independiente que es bajo la supervisión de cardiólogos intervencionistas, EPIC es la única plataforma de este tipo en España, ya que no existe ningún otro foro de registros nacionales que esté financiado por las autoridades sanitarias.
Registros clínicos cardiovasculares: la experiencia belga
Bélgica tiene condiciones muy favorables para la investigación basada en registros al contar con una población de más de 11 millones de personas. Se debe mencionar que es uno de los raros ejemplos donde la recopilación de datos de registros es obligatoria y se financia atendiendo a requisitos nacionales en materia de estándar de cuidados. No obstante, también ejemplifica los desafíos de gobernanza que implica abordar las 3 áreas principales de interés según la definición que da la U.S. Food and Drug Administration: científica, clínica y legislativa. El registro de cardiología intervencionista belga comenzó como una iniciativa impulsada por facultativos para valorar la efectividad de los tratamientos, las disparidades regionales y la adherencia a las guías clínicas a fin de mejorar los resultados de los pacientes y avanzar en el terreno de la investigación científica.
En 1996, el grupo de trabajo belga de cardiología intervencionista comenzó a recopilar un conjunto limitado de datos clínicos y procedimentales (vía fax), sin ningún rigor científico, como es el caso de otros registros de calidad asistencia. Desde 2006, la entrada de datos pasó a una base de datos alojada por la Sociedad Europea de Cardiología (ESC), pero siguió siendo propiedad y gestionada por el grupo de trabajo belga de cardiología intervencionista. En 2012, la base de datos del Registro electrónico de calidad de actos médicos, implantes y dispositivos (Qermid), alojada por las autoridades sanitarias, entró en vigor12. Completar todos los datos es obligatorio para que intervenciones y dispositivos puedan ser reembolsados, lo cual permite recopilar ~100% de todas las intervenciones que se realizan, si bien añade una nueva carga administrativa.
Aunque Qermid recopila datos en materia de legislación y calidad asistencial, en la actualidad, el registro no está liderado ni gestionado por la comunidad científica, lo cual ha generado una paradoja, en virtud de la cual, aunque se da la situación ideal (recopilación completa de datos), no se aprovecha lo suficiente la ventaja científica que da contar con una infraestructura tan valiosa. Tampoco cuenta con recursos dedicados para la entrada de datos online (algo que, en la actualidad, realizan médicos o asistentes) y, por si esto fuera poco, los datos no están suficientemente supervisados. No obstante, gracias al grupo de trabajo belga de cardiología intervencionista y Qermid, Bélgica lleva más de 27 años publicando métricas fiables, proporcionando datos del mundo real a nivel nacional sobre intervenciones complejas13 y describiendo elegantemente los efectos que ha tenido la pandemia de la COVID-1914.
Registros clínicos cardiovasculares: La experiencia sueca
El Registro sueco de angiografía y angioplastia coronaria SCAAR forma parte del sistema web sueco para la mejora y desarrollo de cuidados basados en la evidencia en cardiopatías evaluados según los tratamientos recomendados (SWEDEHEART) y está considerado el modelo a seguir por otros registros coronarios15. Se trata de un registro que fue fundado, a título voluntario, por médicos para mejorar la calidad asistencial. La recopilación de datos se lleva a cabo a nivel nacional, se requiere la entrada de datos para todas las intervenciones y, en la actualidad, el sistema sanitario soporta la infraestructura. No es de extrañar que Suecia maravillara a la comunidad cardiovascular con los primeros ECA basados en registros iniciados por investigadores, publicados en revistas de primer nivel16,17. Con una población de más de 10 millones de personas, los registros de calidad del SWEDEHEART incorporan más de 80.000 intervenciones anuales. La recopilación de datos incluye datos iniciales, de la intervención y de los resultados obtenidos, para un total de más de 300 variables. El registro SWEDEHEART tiene una alta tasa de adherencia por encima del 95% de coincidencia de datos cuando se realizan actividades de seguimiento15.
El registro SWEDEHEART presenta ciertas características que crean condiciones propicias. En primer lugar, todos los pacientes que reciben tratamiento hospitalario están incluidos en el registro. Aunque no se precisa el consentimiento informado del paciente para ingresar a dicho registro, si este decide abandonarlo, se le da la oportunidad de hacerlo. En segundo lugar, se emplea un identificador de paciente común que permite fusionar múltiples bases de datos a nivel nacional, lo cual amplía considerablemente el rango de disponibilidad de datos comparado con un registro hospitalario (tabla 2). Esto permite un seguimiento indefinido a menos que un paciente abandone el país; la infraestructura de gobernanza tiene en cuenta los 3 elementos citados anteriormente (científico, clínico o legislativo); en concreto, el proceso permite enviar feedback al personal, a la dirección, a los pacientes y al público en general.
Tabla 2. Bases de datos conectadas que conforman el SWEDEHEART
| Bases de datos específicas de patologías (por ejemplo, SCAAR para ICP) |
| Registros en la National Board of Health and Welfare |
| El registro nacional de causas de muerte |
| El registro nacional de pacientes (todos los códigos CIE, todos los ingresos desde 1987) |
| El registro sueco de fármacos recetados (todos los fármacos dispensados desde 2005) |
| Central Bureau of Statistics (estado civil, país de nacimiento, ingresos, nivel educativo) |
| La Swedish Social Insurance Agency (gestión de bajas por enfermedad) |
| Otros registros nacionales de calidad (alrededor de 100 en la actualidad) |
|
CIE: Clasificación Internacional de Enfermedades; ICP: intervención coronaria percu- tánea; SCAAR: Registro sueco de angiografía y angioplastia coronaria. |
Desde 2018, los estudios SCAAR/SWEDEHEART se emplean por la industria de dispositivos médicos para avalar informes en materia reguladora en un marco de seguimiento clínico continuo y de completitud de datos. Desde entonces, los estudios SCAAR/SWEDEHEART han avalado a la mayoría de las principales compañías de dispositivos de ICP con informes del MDR. La experiencia actual permite la elaboración de informes predefinidos a nivel de pacientes, dispositivos, subgrupos de pacientes (por ejemplo, adultos mayores, diabéticos) o lesiones (vasos pequeños, lesiones largas). Esta plataforma es especialmente útil para dispositivos que no se utilizan habitualmente (como dispositivos del tronco común izquierdo, stents pequeños). En la actualidad, el modelo SWEDEHEART se implementa en Europa gracias al programa EuroHeart impulsado por la ESC. Algunos países ya contaban con registros similares en diferentes plataformas, por lo que uno de los primeros pasos más importantes fue establecer normas de datos compartidos18.
Registros clínicos cardiovasculares: la experiencia islandesa
La población de Islandia está en torno a las 400.000 personas y cuenta con un centro de cardiología intervencionista en el Landspitali University Hospital (Reikiavik). Este centro viene realizando aportaciones al SCAAR/SWEDEHEART desde 2008 mediante la recopilación prospectiva de datos. Como experiencia unicéntrica y fuera de Suecia, la Dra. Guðmundsdóttir confirma que la recopilación de datos en la base de datos de SCAAR es eficiente en el tiempo y se ve, ya, como parte integral de las historias clínicas de los pacientes. Tanto el médico tratante como las enfermeras de la sala de hemodinámica introducen los datos del paciente y de la intervención en el mismo momento en que se realiza esta, lo cual es preciso y eficiente en el tiempo, dado el abordaje simplificado en materia de recopilación de datos. El registro incluye a todos los pacientes tratados, permite valorar la calidad asistencial y es una vía abierta a la participación en ensayos clínicos multicéntricos basados en registros. El registro permite un acceso fácil a todos los datos islandeses para la investigación de ámbito local y el control de calidad. No obstante, todavía quedan desafíos a los que enfrentarse: a) las bases de datos islandesas no están integradas como en el caso sueco (tabla 2) y b) tanto la entrada de datos como el intercambio de estos tienen cierta complejidad. Como la entrada rutinaria de datos en el registro se considera parte de los cuidados del paciente, no se requiere el consentimiento informado. No obstante, en el caso de estudios basados en registros, sí se requiere la aprobación del comité de ética y la obtención del formulario de consentimiento informado firmado por cada participante. Aunque Islandia colabora en el programa EuroHeart proporcionando datos, en la actualidad, no utiliza la plataforma EuroHeart.
Registros clínicos cardiovasculares: la experiencia de Leiden con un banco de datos de imágenes no invasivas
Leiden University fue fundada en 1575 y es la universidad más antigua de Países Bajos. Su facultad de medicina está muy implicada en innovación y desarrollo y colabora con organizaciones tanto de ámbito local como internacional. Su departamento de cardiología no es una excepción y colabora con 18 países en materia de investigación, programas de doctorado y formación de posgrado. En este contexto, y gracias a una infraestructura y liderazgo muy acertados, en el año 2000 se estableció prospectivamente un potente banco de datos de imágenes no invasivas que ha ido recopilando datos retrospectivos desde 1990. Gracias al uso de protocolos de adquisición estandarizados basados en las trayectorias de asistencia médica (por ejemplo, en la enfermedad o patología del paciente) y esfuerzos de análisis dedicados (impulsados por el infatigable trabajo de becarios de investigación y profesores), Leiden ha logrado ofrecer a la comunidad científica mundial una mejor comprensión de la historia natural de la enfermedad, identificado poblaciones de mayor riesgo e informado sobre el diseño de ensayos clínicos.
La experiencia de Leiden nos deja 5 lecciones importantes: a) cada centro recopila una gran cantidad de datos que, si se utilizan adecuadamente, pueden cambiar nuestra comprensión de la enfermedad y su manejo; b) se requieren metodologías de adquisición y análisis consistentes para la realización de comparativas de datos a lo largo del tiempo. Así, al dedicar tiempo a una buena adquisición de datos, se facilitarán futuros esfuerzos; c) para analizar convenientemente estas enormes bases de datos, se necesitan innumerables horas, lo cual posibilitan programas de doctorado bien organizados; y d) la colaboración entre los registros de imágenes internacionales es especialmente valiosa en el caso de enfermedades menos prevalentes y más productiva cuando se seleccionan buenos centros con un alto volumen de casos, se establecen evaluaciones estandarizadas, las bases de datos están bien organizadas, hay profesionales comprometidos listos para crecer en sus respectivas carreras académicas y la integración de técnicas de imágenes multimodales crea mejores perspectivas para poder responder preguntas clínicamente relevantes. Algunos de estos ejemplos son la experiencia de Leiden con la estenosis aórtica moderada, la valvulopatía aórtica bicúspide y el infarto agudo de miocardio19-21.
Registros clínicos cardiovasculares: perspectiva del European Cardiovascular Research Institute-Cardialysis
El European Cardiovascular Research Institute es una fundación que reúne a una comunidad de destacados investigadores clínicos y socios del sector público/privado para realizar investigaciones clínicas que mejoren la asistencia cardiovascular. Desde 2012, este instituto viene realizando algunos de los ensayos en cardiología intervencionista más ambiciosos de Europa con la inscripción de casi 30.000 participantes y proporcionando datos de alta calidad que han tenido su eco en las guías clínicas de todo el mundo. Como organización de investigación académica que es, el European Cardiovascular Research Institute colabora con Cardialysis, un laboratorio central independiente de imágenes cardiacas orientado a la calidad y organización de investigación centrada en la cardiología. Cardialysis es la organización que más estudios en cardiología intervencionista ha realizado en Europa (más de 400 estudios completados en 40 años y un total de más de 200.000 pacientes inscritos)2. En este marco, desde 2021, Cardialysis viene experimentado una demanda creciente tanto de registros iniciados por la industria como por investigadores en los que es esencial desarrollar cierta concienciación y aceptación común de la calidad requerida para diversos fines tales como aprobación precomercialización, seguimiento poscomercialización, investigación científica, guías… Definir cómo avalar externamente las plataformas de registros con componentes de calidad específicos se ha convertido en una prioridad. El término «registros clínicos avalados externamente» se introdujo en el primer grupo de trabajo del EU-MCVC y alude a redes de registros que utilizan proveedores independientes para mejorar la calidad de los registros en función de los objetivos.
Una llamada a la calidad y participación de las partes interesadas
La cardiología se caracteriza por tener unos estándares muy altos en investigación clínica. La mayoría de las preguntas en materia de investigación en cardiología son comparativas binarias simples. No obstante, la riqueza de información necesaria para planificar una comparativa binaria adecuada acarrea una alta complejidad y requiere de la participación de expertos en varias disciplinas y con conocimientos distintos. Por esta razón, fijar los estándares a seguir se ha convertido en un catalizador efectivo para la innovación. Estos estándares comienzan con los requisitos de las agencias reguladoras3-6 y siguen con las definiciones y principios que dictan el diseño de los ensayos22 pasando por elementos de datos estándar, metodologías estándar y terminan con un proceso estándar de participación de los datos obtenidos. Los ensayos clínicos fallidos que cuentan con las herramientas estadísticas necesarias siguen siendo una característica habitual dentro del contexto de los ensayos clínicos ya que los dispositivos o las estrategias que parecían prometedoras en los ensayos iniciales con un número limitado de pacientes, a veces, tienen datos confirmatorios contradictorios en posteriores ensayos más extensos. Según el criterio del EU-MCVC, los ensayos confirmatorios deben realizarse empleando estándares de alta calidad. Los componentes metodológicos que aportan calidad a una investigación clínica se muestran en la tabla 3.
Tabla 3. Componentes metodológicos que añaden calidad a las investigaciones clínicas cardiovasculares
| Diseño del estudio y desarrollo del protocolo según los estándares internacionales (ISO 14155) |
| Uso de definiciones estándar (definiciones del ARC) |
| Comités de expertos independientes y sin conflicto de intereses (comités directivos, comités de eventos clínicos, juntas de vigilancia de datos y seguridad) |
| Laboratorios centrales de imágenes cardiacas independientes y sin conflictos |
| Selección adecuada de centros (infraestructura de investigación óptima) |
| Vigilancia de centros independiente y sin conflicto de intereses, incluida la verificación de datos (integridad, precisión) |
| Codificación consistente de las reacciones adversas (MedDRA) |
| Sistema electrónico de recopilación de datos conforme a la legislación vigente |
| Plan de análisis estadístico y estrategia de publicación predefinida |
| Informes estadísticos independientes o validación estadística independiente |
| Uso adecuado en tiempo y forma de bases de datos públicas (ClinicalTrials.gov) |
| Garantía consistente de calidad en cumplimiento de la legislación vigente y auditorías de los centros |
| Informes de estudios clínicos conforme a estándares internacionales (ISO 14155) |
|
ARC: Academic Research Consortium; ISO: Organización Internacional de Normalización; MedDRA: Diccionario Médico para Actividades Reguladoras. |
En una reciente revisión sistemática, Coordinating Research and Evidence for Medical Devices (CORE-MD), publicó los resultados de su estudio de los 11 registros europeos que existen en la actualidad, sobre stents coronarios y terapias valvulares percutáneas23. Concluyeron que existen una amplia heterogeneidad y una incompleta transparencia pública tanto en la estructura como en los métodos, así como una necesidad de crear un conjunto mínimo de criterios de calidad. Informaron que, de media, los criterios de calidad y completitud de los datos se cumplían en menos del 20% de los registros y que los datos de seguridad y rendimiento se abordaban como es debido en menos del 30%. Esta información viene a confirmar que la prioridad sigue siendo mejorar la calidad de la recopilación de datos y que se deben desarrollar métricas ampliamente aceptadas.
Algo que requiere más investigación es la necesidad y el valor relativo de las actividades de seguimiento y auditorías in situ en el contexto de los registros clínicos. Aunque los mecanismos automatizados y centralizados de seguimiento de datos pueden fomentar la eficiencia, todavía se desconoce cuál es el efecto que tienen las visitas de seguimiento in situ sobre la completitud y calidad de los datos. En líneas generales, el seguimiento in situ se viene empleando en registros de dispositivos impulsados por patrocinadores, no así en registros de pacientes impulsados por la comunidad académica.
Mejoras en la calidad de los registros clínicos tradicionales
Análisis independientes en laboratorios centrales
Establecer un laboratorio central independiente para un ensayo clínico aumenta la calidad abordando los siguientes requisitos de calidad: a) optimizando la calidad de las imágenes mediante el desarrollo de un protocolo uniforme de obtención de imágenes para todos los centros participantes. La adherencia al protocolo de obtención podría requerir confirmación de que se estudió (o recibió formación) el protocolo de obtención de imágenes y se proporcionó una prueba para confirmar el cumplimiento de este; b) garantizando que los datos se traten de manera consistente (por ejemplo, a través de seudonimización, estándares adecuados de privacidad y seguridad, un formato adecuado, software de análisis consistente); c) garantizando que los datos se analicen de consistentemente (por ejemplo, a través de metodología estándar, valoraciones reproducibles, personal debidamente capacitado); d) garantizando que la adjudicación de datos sea consistente (por ejemplo, la gravedad de la insuficiencia) y e) disponibilidad central de los conjuntos de datos originales para auditorías regulatorias24.
Tanto las mediciones como las valoraciones de las imágenes obtenidas a partir de datos del mundo real (por ejemplo, facilitadas in situ) no cumplirán los requisitos de calidad mencionados en el párrafo anterior al estar asociados a una mayor variabilidad en las valoraciones, así como a un mayor riesgo de sesgo por parte del investigador. A efectos de la investigación académica, pueden abordarse los requisitos 1 y 4 señalados en el párrafo anterior siendo aceptable la ausencia del resto siempre y cuando los datos de imágenes no salgan del centro tratante. No obstante, a efectos de los ensayos regulatorios, los 5 requisitos son necesarios, sobre todo, si las imágenes son parte del objetivo primario o del principal mecanismo de acción del dispositivo en investigación. En el contexto de los registros clínicos poscomercialización, dado el gran número potencial de pacientes, se deben diseñar soluciones intermedias. La tabla 4 ilustra las diferencias entre un laboratorio central sujeto que cumple con la legislación vigente y un laboratorio central académico de ámbito local.
Tabla 4. Requisitos para laboratorios de imágenes académicos frente a los que cumplen con la legislación vigente
| Laboratorio de imágenes que cumple con la legislación vigentea | Laboratorio de imágenes académico de ámbito local | |
|---|---|---|
| Obtención de imágenes | ||
| Manual/protocolo de imágenes específico del estudio | Sí | No/Sía |
| Capacitación/calificación específica del personal del estudio | Sí | No/Sía |
| Recursos destinados a la obtención de imágenes | Sí | No/Sía |
| Gestión de los datos proporcionados por las imágenes | ||
| Anonimización de Información de Salud Protegida | Sí | Noc |
| Sistema seguro de transferencia electrónica de imágenes | Sí | Noc |
| Manejo adecuado del material y archivo | Sí | Noc |
| Manejo del feedback en materia de calidad y consultas | Sí | Noc |
| Análisis de imágenes | ||
| Metodología de análisis estandarizada (de conformidad con las guías, definiciones aceptadas, garantizando la viabilidad) | Sí | No/Síb |
| Estación de trabajo de imágenes dedicada | Sí | No/Síb |
| Lector principal - ecografista/analista de imágenes | Sí | No/Síb |
| Lectura adicional - experto/supervisor de imágenes | Sí | No/Síb |
| Capacitación/formación específica del personal | Sí | No |
| Pruebas de reproducibilidad | Sí | No |
| Base de datos de imágenes | ||
| Formulario electrónico de casos específico del estudio validado | Sí | No |
| Altos requisitos de entrada de datos (carga automática de hojas de cálculo y consultas, cumplimiento de la Parte 11, registro de auditoría) | Sí | No |
| Verificación de fuente de datos y control de calidad | Sí | No |
| Procedimiento de liberación de datos tras el cierre de la base de datos | Sí | No |
|
a Pamela Douglas JASE. b No en registros, pero posible en estudios académicos. c No aplicable a un estudio de ámbito local. |
||
Adjudicación independiente de objetivos
Contar con un comité de eventos clínicos (CEC) independiente aumenta la calidad al abordar los siguientes requisitos de calidad: a) adherencia a definiciones estándar para determinar y clasificar eventos adversos que potencialmente cumplen con la definición de objetivo de un determinado estudio. Contar con un comité de expertos en un ensayo también otorga consistencia a la hora de clasificar eventos complejos tales como infarto de miocardio perioperatorios e insuficiencias cardiacas; b) garantizar que las valoraciones se realicen empleando una cantidad similar de información (por ejemplo, una lista de verificación consistente de documentos y materiales gráficos necesarios a efectos de adjudicación); c) disponibilidad central de documentos fuente originales para auditorías regulatorias; y d) como las indicaciones de los dispositivos se basan, en gran medida, en objetivos primarios clínicos, es importante que el CEC demuestre ser un órgano independiente del fabricante de un determinado dispositivo y no tener ningún conflicto de intereses que pueda lastrarle a la hora de cabo las tareas consignadas25.
En el contexto de la aprobación precomercialización, el EU-MCVC considera que la presencia de un CEC independiente es necesaria por las 4 razones expuestas en el párrafo anterior. No obstante, en el marco de un registro cardiovascular, parece que los datos informados in situ, sobre todo, cuando el informe es completo y emplea definiciones estándar, podrían bastar desde una perspectiva de calidad. Desde el punto de vista científico, aún está por demostrar si los datos de resultados clínicos de los registros son suficientes sin el concurso de un CEC. Además, en el actual contexto de estudios aleatorizados basados en registros en materia regulatoria, en opinión del EU-MCVC, es necesario contar con un CEC y su uso y validez deberían explorarse prospectivamente.
En cualquier caso, la mortalidad por cualquier causa no suele necesitar adjudicación de objetivos, sobre todo, si el registro o estudio específico tiene acceso a bases de datos nacionales sobre mortalidad. Se desconoce si las subclasificaciones de muerte podrían documentarse de manera confiable utilizando datos informados in situ o si un CEC aporta un valor adicional. Otros objetivos sobre los cuales se está trabajando para aclarar si la adjudicación es o no beneficiosa son el infarto de miocardio y la revascularización, cuando se codifican como binarios (sí/no). En opinión del EU-MCVC, casi todos los demás objetivos (insuficiencia cardíaca, sangrado, subtipos de infarto de miocardio, trombosis del stent, accidente cerebrovascular, angina inestable, revascularización no programada) muestran una ventaja cuando se someten a adjudicación, aunque todavía hay que demostrar esto prospectivamente.
Un ejemplo de adjudicación de eventos clínicos en un ECA basado en registros es el estudio VALIDATE SWEDEHEART (Bivalirudina frente a monoterapia con heparina en infarto de miocardio), que fue una prueba de concepto de esta metodología26. También se están diseñando y probando innovadores abordajes de adjudicación para mantener la calidad y reducir costes. Por ejemplo, en el estudio DAPA-MI, solo se adjudicaron la muerte y la hospitalización por insuficiencia cardiaca, mientras que el infarto de miocardio, la revascularización y el accidente cerebrovascular se informaron in situ27. Se presentan ejemplos adicionales en la tabla 5.
Tabla 5. Uso de la adjudicación de objetivos en ensayos clínicos aleatorizados basados en registros
| Estudio | Objetivo | Adjudicación | Objetivos del registro | Disparador del evento | Recopilación de datos | Otra información |
|---|---|---|---|---|---|---|
| TASTE | Muerte por cualquier causa | No | Sí | ND | No | - |
| VALIDATE | MACE y sangrado mayor | Sí | ND | Informado por el propio centro | Sí, FECC y notas del hospital | - |
| DETOX | Muerte por cualquier causa | No | Sí | ND | No | - |
| iFR | MACE y sangrado mayor | Sí | ND | Informado por el propio centro | Sí, FECC y notas del hospital | Laboratorio central |
| HELP | Episodios hemorrágicos | ND | Sí | ND | Sí | - |
| REDUCE | Muerte por cualquier causa e IM | ND | Sí | ND | No | - |
| Full REVASC | IM y revascularización no programada | Sí | ND | SCAAR/Riks-HIA | Sí, FECC y notas del hospital | - |
| SPIRRIT | Muerte por cualquier causa y hospitalización por IC | Sí | Códigos ICD | Códigos ICD y registro de mortalidad | Sí, FECC y notas del hospital | Proceso de adjudicación simplificado |
| DAPA-MI | Muerte por cualquier causa y hospitalización por IC | Sí | ND | Informado por el propio centro | Sí, FECC y notas del hospital | - |
| INFINITY | Objetivo combinado orientado al dispositivo | Sí | ND | Informado por el propio centro | Sí, FECC y notas del hospital | Laboratorio central |
|
FECC: formulario electrónico de registro de casos clínicos; IC: insuficiencia cardiaca; ICD: Clasificación Internacional de Enfermedades; IM: infarto de miocardio; MACE: eventos cardiovasculares adversos mayores; Riks-HIA: Registro sueco de información y conocimiento sobre ingresos en unidades de cuidados intensivos cardiacos de Suecia; SCAAR: Registro sueco de angiografía y angioplastia coronaria. |
||||||
Análisis o validación estadística independiente
El estudio de bases de datos de ensayos apropiadas va más allá de la base de datos de análisis estadístico bloqueada e incluye una valoración exhaustiva del diseño de un ensayo o registro. Desde la perspectiva de un experto en estadística, se debe tener en cuenta el diseño del estudio, la selección de pacientes, la elección del comparador, el cumplimiento normativo, la descripción de los méto- dos estadísticos y, por último, tanto la valoración de los resultados per se como la consistencia de los hallazgos. La brecha de calidad de los datos entre el paradigma de la medicina basada en la evidencia y el paradigma de los datos del mundo real es evidente en la actualidad y esto es conceptualmente correcto por diseño, dado que los datos del mundo real aluden a datos recopilados rutinariamente diseñados con una menor granularidad y precisión que los datos de los ensayos clínicos. Cuando los datos del mundo real se usan a efectos regulatorios y normativos, estos deben cumplir los siguientes requisitos: a) las fuentes de datos han de ser de buena calidad; b) se espera validez interna y externa; c) consistencia entre las fuentes de datos; y d) los datos deberán ser adecuados y precisos. Los documentos en materia regulatoria que emplean datos del mundo real también deben informar sobre el ajuste de posibles factores de confusión, identificar los posibles sesgos de selección e información, describir cuál debe ser el manejo de los datos faltantes y ofrecer un análisis de datos robusto.
Los registros clínicos adecuadamente diseñados y avalados son muy ventajosos desde un punto de vista clínico, pues ofrecen una mejor comprensión de la historia natural de las enfermedades, una mejor caracterización de las poblaciones diana y la identificación de nuevas dianas terapéuticas. Los registros también ofrecen la posibilidad de introducir nuevos abordajes estadísticos para agrupar y analizar datos. Cuando hay datos de pacientes disponibles en un único registro, se deben emplear abordajes estadísticos tradicionales, teniendo en cuenta las limitaciones de los datos. Cuando solo hay disponibles datos de registro, se pueden implementar métodos meta-analíticos que pueden usarse en materia legislativa o para la toma de decisiones de salud pública, pero no para valorar la seguridad, eficacia ni efectividad de un dispositivo, algo que requiere la mayor granularidad de todas y que no proporcionan los datos meta-analíticos del registro.
Papel que juegan los registros clínicos en los comités europeos en materia de guías clínicas
El proceso de generación de evidencia que conduce a las recomendaciones de las guías clínicas europeas está bien establecido y sigue los estándares más sólidos. En este proceso, los ensayos clínicos aleatorizados controlados y dotados de las herramientas estadísticas necesarias son la mejor fuente de información para la toma de decisiones. En el mejor de los casos, una recomendación clase IA debería contar con más de un ensayo clínico confirmatorio adecuadamente potenciado o, como mínimo, un metanálisis realizado correctamente. A falta de ECA, no obstante, se emplean otras fuentes de datos que, en última instancia, contribuyen al proceso de toma de decisiones de un comité.
Con el objetivo de optimizar tanto la asistencia clínica como los resultados cardiovasculares, la ESC ha propuesto un abordaje metódico para el desarrollo de indicadores de calidad y, en colaboración con los registros europeos unificados para la evaluación de los cuidados cardiacos y posteriores ensayos aleatorizados (EuroHeart), ha propuesto una serie de estándares de datos para el síndrome coronario agudo y la ICP, el implante percutáneo de válvula aórtica, la insuficiencia cardiaca y la fibrilación auricular/ablación con catéter del flúter18. Se han adoptado rápidamente con la incorporación de 40.000 casos provenientes de una serie de datos individuales de participantes recopilados en 2022 en 7 países participantes. Este nuevo ecosistema está evolucionando rápidamente y promete dejar huella tanto en materia legislativa como en las prioridades de salud pública.
Las fortalezas existentes de los registros clínicos convenientemente planificados y avalados incluyen ser efectivos en recursos, ofrecer una alta representatividad, integrarse en la rutina clínica y un reclutamiento consecutivo no seleccionado. Este contexto, sobre todo tal y como lo ha implementado el SWEDEHEART, ha abonado el camino para ECA basados en registros que reducen la carga de trabajo, minimizan el sesgo seleccionado, brindan un mejor acceso para poder acometer investigaciones impulsadas por los propios investigadores y, recientemente, han abierto la puerta a la posibilidad de realizar ensayos multinacionales. La investigación continuada puede informar mejor tanto a la comunidad científica como a los comités que elaboran guías clínicas sobre el uso de los resultados de ECA basados en registros para la toma de decisiones, siendo su papel final la medicina basada en la evidencia. La tabla 6 ofrece una comparativa entre registros tradicionales, ECA basados en registros y ECA.
Tabla 6. Tabla comparativa sobre el papel que juegan los registros clínicos en la medicina basada en la evidencia
| Registros | Ensayos controlados aleatorizados basados en registros | Ensayos controlados aleatorizados tradicionales |
|---|---|---|
| Meramente observacionales No son aptos para avalar una conclusión sobre eficacia en materia científica |
Pragmáticos Valoración abierta de opciones terapéuticas habitualmente utilizadas |
Máximo nivel de evidencia científica Patrón oro para estudios comparativos |
| De pacientes no seleccionados – representatividad Proporcionan datos para calcular la potencia estadística Resultados significativos a nivel clínico Eventos poco habituales |
De pacientes no seleccionados – representatividad Proporciona información sobre las características y el seguimiento de los pacientes |
Selección de pacientes elegibles Obtención del consentimiento informado Aleatorización del tratamiento Control de los factores de confusión Detección y adjudicación de objetivos clínicos |
| Generadores de hipótesis | Inferencia causal Para valoración de tratamientos, estrategias o agentes farmacológicos en fase aguda Valoración de agentes farmacológicos para nuevas indicaciones |
Inferencia causal |
| Eficientes en recursos | Bajo coste | Intensos en recursos |
Limitaciones
La información presentada y las organizaciones representadas en este manuscrito se limitan a la experiencia de los participantes del think tank que se reunió en Róterdam el pasado 8 de septiembre de 2023 y describe las perspectivas de los coautores. Este no es un documento de consenso ni una revisión sistemática. No se recopiló ni representó información sobre otras organizaciones o países implicados en el desarrollo ni en la elaboración de registros de cardiología intervencionista. El EU-MCVC da la bienvenida a la participación voluntaria de otras organizaciones de investigación cardiovascular establecidas o sociedades de cardiología. La vía de contacto con el EU-MCVC es el autor para correspondencia.
CONCLUSIONES
El EU-MDR ha ido incrementando los requisitos en lo referente a las actividades de seguimiento poscomercialización que deben cumplir todos los fabricantes de dispositivos que comercializan dispositivos médicos en Europa. Los registros clínicos adecuadamente planificados y avalados tienen el potencial de abordar requisitos adicionales a través de la creación de marcos de colaboración. La calidad de los datos es una preocupación esencial y tanto los registros actuales como las futuras plataformas de registro deben incluir estrategias para optimizar la calidad de los datos a fin de cumplir con los requisitos en materia regulatoria. Este abordaje colaborativo tiene el potencial de mejorar la calidad asistencial, reducir los costes y permitir un acceso más rápido a tecnologías innovadoras. Los registros, redes, estándares e intervenciones existentes deben ser adoptados y empleados consistentemente. Los múltiples usos potenciales de la recopilación de datos basada en registros hacen que sea un área que merece una mayor y continua atención por todas las partes interesadas de la industria en materia de dispositivos médicos, con el objetivo conjunto de mejorar tanto el manejo como los resultados cardiovasculares.
FINANCIACIÓN
Ninguna.
DECLARACIÓN SOBRE EL USO DE INTELIGENCIA ARTIFICIAL
No utilizada.
CONTRIBUCIÓN DE LOS AUTORES
E. Spitzer y J.G.P. Tijssen contribuyeron al inicio, diseño y planificación del estudio. E. Spitzer, J.M. de la Torre Hernández, I.J. Guðmundsdóttir, E. McFadden, C. Held, C. Hanet, E. Boersma, C.B. Ren, V. Delgado, D. Erlinge, A. Pérez de Prado, J. Bax y J.G.P. Tijssen contribuyeron aportando datos/presentaciones. E. Spitzer compiló y redactó el manuscrito cuya revisión y corrección corrió a cargo de J.M. de la Torre Hernández, I.J. Guðmundsdóttir, E. McFadden, C. Held, C. Hanet, E. Boersma, C.B. Ren, V. Delgado, D. Erlinge, A. Pérez de Prado, J. Bax y J.G.P. Tijssen. Todos los autores aprobaron la versión final del manuscrito.
CONFLICTO DE INTERESES
E. Spitzer declaró haber suscrito contratos institucionales por los que no recibe compensación directa con Boston Scientific, Cardiawave, Edwards Lifesciences, Medtronic, Shanghai Microport Medical Co Ltd, NVT GmBH, Pie Medical Imaging y Siemens Healthcare GmBH. C.B. Ren declaró haber suscrito contratos institucionales para la realización de análisis en laboratorios de ecocardiografía con Boston Scientific, Cardiawave, Edwards Lifesciences, NVT GmBH/Biosensores, por los que no ha recibido ninguna compensación personal. Por otro lado, declaró haber recibido honorarios como conferenciante de Abbott. V. Delgado declaró haber recibido honorarios como conferenciante de Edwards Lifesciences, GE Healthcare, Novartis y Philips y consultor de Edwards Lifesciences, Novo Nordisk y MSD. A. Pérez de Prado es presidente de Fundación EPIC. J.G.P. Tijssen es miembro de la junta directiva del European Cardiovascular Research Institute. Los demás autores no declararon ningún otro conflicto de intereses.
A. Pérez de Prado es editor asociado de REC: Interventional Cardiology. Se ha seguido el procedimiento editorial establecido en la revista para garantizar la gestión imparcial del manuscrito.
J.M. de la Torre Hernández es editor jefe de REC: Interventional Cardiology. Se ha seguido el procedimiento editorial establecido en la revista para garantizar la gestión imparcial del manuscrito.
REFERENCES
1. European Parliament and the Council of the European Union. Regulation (EU) 2017/745 of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC. Disponible en: https://eur-lex.europa.eu/eli/reg/2017/745/oj. Consultado 9 Ene 2024.
2. Spitzer E. Forty years of Cardialysis:a leading European cardiovascular research organization. REC:Interv Cardiol. 2023;5:250-253.
3. US Food &Drud Administration (FDA). Use of Real-World Evidence to Support Regulatory Decision-Making for Medical Devices:Guidance for Industry and Food and Drug Administration Staff. 2017. Disponible en: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-real-world-evidence-support-regulatory-decision-making-medical-devices. Consultado 9 Ene 2024.
4. European Medicines Agency (EMA). Patient Registry Initiative - Strategy and Mandate of the Cross-Committee Task Force. 2017. Disponible en: https://www.ema.europa.eu/en/documents/other/patient-registry-initiative-strategy-mandate-cross-committee-task-force_en.pdf. Consultado 9 Ene 2024.
5. European Medicines Agency (EMA). Guideline on Registry-based Studies. 2021. Disponible en: https://www.ema.europa.eu/en/guideline-registry-based-studies-scientific-guideline. Consultado 9 Ene 2024.
6. International Medical Device Regulators Forum (IMDRF). Tools for Assessing the Usability of Registries in Support of Regulatory Decision-Making. 2018. Disponible en: https://www.imdrf.org/documents/tools-assessing-usability-registries-support-regulatory-decision-making. Consultado 9 Ene 2024.
7. de Boer SP, Lenzen MJ, Oemrawsingh RM, et al. Evaluating the 'all-comers'design:a comparison of participants in two 'all-comers'PCI trials with non-participants. Eur Heart J. 2011;32:2161-2167.
8. Jurado-Roman A, Freixa X, Cid B, et al. Spanish cardiac catheterization and coronary intervention registry. 32nd official report of the Interventional Cardiology Association of the Spanish Society of Cardiology (1990-2022). Rev Esp Cardiol. 2023;76:1021-1031.
9. Biswas S, Lefkovits J, Liew D, Gale CP, Reid CM, Stub D. Characteristics of national and major regional percutaneous coronary intervention registries:a structured literature review. EuroIntervention. 2018;14:1112-1120.
10. Freixa X, Jurado-Roman A, Cid B, Cruz-Gonzalez I. Spanish cardiac catheterization and coronary intervention registry. 31st official report of the Interventional Cardiology Association of the Spanish Society of Cardiology (1990-2021). Rev Esp Cardiol. 2022;75:1040-1049.
11. de la Torre-Hernández JM, Alfonso F, Hernandez F, et al. Drug-eluting stent thrombosis:results from the multicenter Spanish registry ESTROFA (Estudio ESpanol sobre TROmbosis de stents FArmacoactivos). J Am Coll Cardiol. 2008;51:986-90.
12. Hanet C, Claeys MJ, Carlier M, Desmet W. Quality assessment in percutaneous coronary interventions:the QERMID Belgian PCI registry. Acta Cardiol. 2018;73:388-391.
13. Kayaert P, Coeman M, Demolder A, et al. Mortality in STEMI Patients With Cardiogenic Shock:Results From a Nationwide PCI Registry and Focus on Left Main PCI. J Invasive Cardiol. 2022;34:E142-E148.
14. Claeys MJ, Argacha JF, Collart P, et al. Impact of COVID-19-related public containment measures on the ST elevation myocardial infarction epidemic in Belgium:a nationwide, serial, cross-sectional study. Acta Cardiol. 2021;76:863-869.
15. Jernberg T, Attebring MF, Hambraeus K, et al. The Swedish Web-system for enhancement and development of evidence-based care in heart disease evaluated according to recommended therapies (SWEDEHEART). Heart. 2010;96:1617-21.
16. Erlinge D, Omerovic E, Frobert O, et al. Bivalirudin versus Heparin Monotherapy in Myocardial Infarction. N Engl J Med. 2017;377:1132-1142.
17. Frobert O, Lagerqvist B, Olivecrona GK, et al. Thrombus aspiration during ST-segment elevation myocardial infarction. N Engl J Med. 2013;369:1587-1597.
18. Batra G, Aktaa S, Wallentin L, et al. Methodology for the development of international clinical data standards for common cardiovascular conditions:European Unified Registries for Heart Care Evaluation and Randomised Trials (EuroHeart). Eur Heart J Qual Care Clin Outcomes. 2023;9:161-168.
19. Butcher SC, Prevedello F, Fortuni F, et al. Prevalence and Prognostic Implications of Moderate or Severe Mitral Regurgitation in Patients with Bicuspid Aortic Valve. J Am Soc Echocardiogr. 2023;36:402-410.
20. Laenens D, Stassen J, Galloo X, et al. The impact of atrial fibrillation on prognosis in aortic stenosis. Eur Heart J Qual Care Clin Outcomes. 2023;9:778-784.
21. Nabeta T, Meucci MC, Westenberg JJM, et al. Prognostic implications of left ventricular inward displacement assessed by cardiac magnetic resonance imaging in patients with myocardial infarction. Int J Cardiovasc Imaging. 2023;39:1525-1533.
22. Spitzer E, McFadden E, Vranckx P, et al. Critical Appraisal of Contemporary Clinical Endpoint Definitions in Coronary Intervention Trials:A Guidance Document. JACC Cardiovasc Interv. 2019;12:805-819.
23. Hoogervorst LA, Geurkink TH, Lubbeke A, et al. Quality and Utility of European Cardiovascular and Orthopaedic Registries for the Regulatory Evaluation of Medical Device Safety and Performance Across the Implant Lifecycle:A Systematic Review. Int J Health Policy Manag. 2023;12:7648.
24. Douglas PS, DeCara JM, Devereux RB, et al. Echocardiographic imaging in clinical trials:American Society of Echocardiography Standards for echocardiography core laboratories:endorsed by the American College of Cardiology Foundation. J Am Soc Echocardiogr. 2009;22:755-765.
25. Spitzer E, Fanaroff AC, Gibson CM, et al. Independence of clinical events committees:A consensus statement from clinical research organizations. Am Heart J. 2022;248:120-129.
26. Erlinge D, Koul S, Eriksson P, et al. Bivalirudin versus heparin in non-ST and ST-segment elevation myocardial infarction-a registry-based randomized clinical trial in the SWEDEHEART registry (the VALIDATE-SWEDEHEART trial). Am Heart J. 2016;175:36-46.
27. James S, Erlinge D, Storey RF, et al. Rationale and design of the DAPA-MI trial:Dapagliflozin in patients without diabetes mellitus with acute myocardial infarction. Am Heart J. 2023;266:188-197.
